



Next: Performance Tuning
Up: Runtime Analysis
Previous: Pair interaction calculations
Contents
Index
NAMD supports the calculation of lateral pressure profiles as a function of
the z-coordinate in the system. The algorithm is based on that of
Lindahl and Edholm (JCP 2000), with modifications to enable Ewald sums based on
Sonne et al (JCP 122, 2005).
The simulation space is partitioned into slabs, and half the virial
due to the interaction between two particles is assigned to each
of the slabs containing the particles. This amounts to employing
the Harasima contour, rather than the Irving-Kirkwood contour, as
was done in NAMD 2.5. The diagonal components of the pressure
tensor for each slab, averaged over all timesteps since the previous
output, are recorded in the NAMD output file. The
units of pressure are the same as in the regular NAMD pressure
output; i.e., bar.
The total virial contains contributions from up to four components:
kinetic energy, bonded interactions, nonbonded interactions, and an Ewald
sum. All but the Ewald sums are computed online during a normal simulation
run (this is a change from NAMD 2.5, when nonbonded contributions to the
Ewald sum were always computed offline). If the simulations are performed
using PME, the Ewald contribution should be estimated using a separate,
offline calculation based on the saved trajectory files. The nonbonded
contribution using a cutoff different from the one used in the simulation
may also be computed offline in the same fashion as for Ewald, if desired.
Pressure profile calculations may be performed in either constant volume
or constant pressure conditions. If constant pressure is enabled, the
slabs thickness will be rescaled along with the unit cell; the dcdUnitCell
option will also be switched on so that unit cell information is stored in
the trajectory file.
NAMD 2.6 now reports the lateral pressure partitioned by interaction type.
Three groups are reported: kinetic + rigid bond restraints (referred to as
``internal", bonded, and nonbonded. If Ewald pressure profile calculations
are active, the Ewald contribution is reported in the nonbonded section, and
no other contributions are reported.
NAMD 2.6 also permits the pressure profile to be partitioned by atom type.
Up to 15 atom groups may be assigned, and individual contribution of each
group (for the ``internal" pressures) and the pairwise contributions of
interactions within and between groups (for the nonbonded and bonded pressures)
are reported in the output file.
- pressureProfile
 compute pressure profile
compute pressure profile 
Acceptable Values: on or off
Default Value: off
Description:
When active, NAMD will compute kinetic, bonded and nonbonded (but not
reciprocal space) contributions to the
pressure profile. Results will be recorded in the NAMD output file
in lines with the format
PRESSUREPROFILE: ts Axx Ayy Azz Bxx Byy Bzz ... , where ts is the
timestep, followed by the three diagonal components of the pressure tensor
in the first
slab (the slab with lowest z), then the next lowest slab, and so forth.
The output will reflect the pressure profile averaged over all the steps since
the last output.
NAMD also reports kinetic, bonded and nonbonded contributions separately,
using the same format as the total pressure, but on lines beginning with
PPROFILEINTERNAL, PPROFILEBONDED, and PPROFILENONBONDED.
- pressureProfileSlabs
Number of slabs in the spatial partition
Acceptable Values: Positive integer
Default Value: 10
Description:
NAMD divides the entire periodic cell into horizontal slabs of equal
thickness; pressureProfileSlabs specifies the number of such slabs.
- pressureProfileFreq
How often to output pressure profile
data
Acceptable Values: Positive integer
Default Value: 1
Description:
Specifies the number of timesteps between output of pressure profile data.
- pressureProfileEwald
Enable pressure profile Ewald sums
Acceptable Values: on or off
Default Value: off
Description:
When enabled, only the Ewald contribution to the pressure profile will be
computed. For trajectory analysis the
recommended way to use this option is to use the NAMD Tcl scripting
interface as described in Sec. 2.2.2 to run for
0 steps, so that NAMD prints the pressure profile without performing any
dynamics.
The Ewald sum method is as described in Sonne et al. (JCP 122, 2005). The
number of  vectors to use along each periodic cell dimension is specified
by the pressureProfileEwald
vectors to use along each periodic cell dimension is specified
by the pressureProfileEwald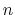 parameters described below.
parameters described below.
- pressureProfileEwaldX
Ewald grid size along X
Acceptable Values: Positive integer
Default Value: 10
Description:
- pressureProfileEwaldY
Ewald grid size along Y
Acceptable Values: Positive integer
Default Value: 10
Description:
- pressureProfileEwaldZ
Ewald grid size along Z
Acceptable Values: Positive integer
Default Value: 10
Description:
- pressureProfileAtomTypes
Number of atom type partitions
Acceptable Values: Positive integer
Default Value: 1
Description:
If pressureProfileAtomTypes is greater than 1, NAMD will calculate
the separate contributions of each type of atom to the internal, bonded,
nonbonded, and total pressure. In the case of the internal contribution,
there will be
pressure profile data sets reported on each
PPROFILEINTERNAL line, where
is the number of atom types. All the
partial pressures for atom type 1 will be followed by those for atom type 2,
and so forth. The other three pressure profile reports will contain
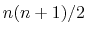 data sets. For example, if there are
data sets. For example, if there are 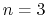 atom types, the
six data sets arising from the three inter-partition and the three
intra-partition interactions will be reported in the following order:
1-1, 1-2, 1-3, 2-2, 2-3, 3-3. The total pressure profile, reported
on the PRESSUREPROFILE line, will contain the internal contributions
in the data sets corresponding to 1-1, 2-2, etc.
atom types, the
six data sets arising from the three inter-partition and the three
intra-partition interactions will be reported in the following order:
1-1, 1-2, 1-3, 2-2, 2-3, 3-3. The total pressure profile, reported
on the PRESSUREPROFILE line, will contain the internal contributions
in the data sets corresponding to 1-1, 2-2, etc.
- pressureProfileAtomTypesFile
Atom type partition assignments
Acceptable Values: PDB file
Default Value: coordinate file
Description:
If pressureProfileAtomTypes is greater than 1, NAMD will assign
atoms to types based on the corresponding value in pressureProfileAtomTypesCol. The type for each atom must be strictly less than
pressureProfileAtomTypes!
- pressureProfileAtomTypesCol
pressureProfileAtomTypesFile
PDB column
Acceptable Values: PDB file
Default Value: B
Description:
Here is an example snippet from a NAMD input that can be used to compute
the Ewald component of the pressure profile. It assumes that the
coordinates were saved in the dcd file pp03.dcd) every 500 timesteps.
Pme on
PmeGridSizeX 64
PmeGridSizeY 64
PmeGridSizeZ 64
exclude scaled1-4
1-4scaling 1.0
switching on
switchdist 9
cutoff 10
pairlistdist 11
pressureProfile on
pressureProfileSlabs 30
pressureProfileFreq 100
pressureProfileAtomTypes 6
pressureProfileAtomTypesFile atomtypes.pdb
pressureProfileEwald on
pressureProfileEwaldX 16
pressureProfileEwaldY 16
pressureProfileEwaldZ 16
set ts 0
firstTimestep $ts
coorfile open dcd pp03.dcd
while { [coorfile read] != -1 } {
incr ts 500
firstTimestep $ts
run 0
}
coorfile close
Next: Performance Tuning
Up: Runtime Analysis
Previous: Pair interaction calculations
Contents
Index
http://www.ks.uiuc.edu/Research/namd/