From: Martin, Erik W (Erik.Martin_at_stjude.org)
Date: Wed Oct 24 2012 - 14:33:41 CDT
Yeah, I'm definitely confused as well. The conf file I attached was from after I'd turned rigid bonds off… not sure why I changed the flag to "none" instead of just commenting it out. Regardless, I was at a stage where I didn't want my water to be rigid. I've never used Tip4p before this, does it commonly have problems if you turn rigid bonds off?
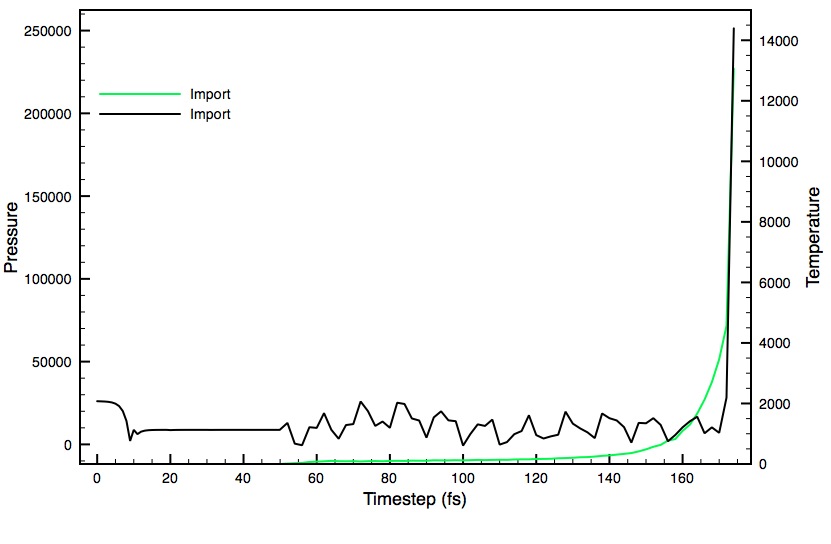
To answer your other question, the atoms that are moving too fast are more often water, but I think that's purely because there are more waters atoms in my simulation than peptide. I'll attach a plot of the temperature and pressure from the last crash I saw so you can see what I mean. It seems that the temperature jumps causing a pressure increase and clearly at that temp something is going to start moving too fast (I'd think).
Funnily enough, the simulation that plot was from was supposed to be a slow heating simulation but ended up not working (I'll attach that configuration file too)
Thanks,
Erik
From: Aron Broom <broomsday_at_gmail.com<mailto:broomsday_at_gmail.com>>
Date: Wed, 24 Oct 2012 12:56:36 -0500
To: Erik Martin <erik.martin_at_stjude.org<mailto:erik.martin_at_stjude.org>>
Cc: "namd-l_at_ks.uiuc.edu<mailto:namd-l_at_ks.uiuc.edu>" <namd-l_at_ks.uiuc.edu<mailto:namd-l_at_ks.uiuc.edu>>
Subject: Re: namd-l: unreasonably high temp/pressure
hmm, I'm confused. Why would you have "rigidbonds none" for TIP4P instead of "rigidbonds water"? Other than that I can't see anything weird.
Have you tried slowly heating? Maybe more minimization after the equilibration? You said it's different atoms each time that cause the problem, but are they usually peptide atoms or water atoms?
On Wed, Oct 24, 2012 at 11:34 AM, Martin, Erik W <Erik.Martin_at_stjude.org<mailto:Erik.Martin_at_stjude.org>> wrote:
Yep. I did set that. It does seem to have something to do with the water though given that the problem arises when I turn rigid bonds off. If it helps, I'll attach one of my configuration files… There might be some odd values there though given that I started changing some things to try to rectify the problem (margin and cutoff distances etc)
From: Aron Broom <broomsday_at_gmail.com<mailto:broomsday_at_gmail.com><mailto:broomsday_at_gmail.com<mailto:broomsday_at_gmail.com>>>
Date: Wed, 24 Oct 2012 10:26:43 -0500
To: Erik Martin <erik.martin_at_stjude.org<mailto:erik.martin_at_stjude.org><mailto:erik.martin_at_stjude.org<mailto:erik.martin_at_stjude.org>>>
Cc: "namd-l_at_ks.uiuc.edu<mailto:namd-l_at_ks.uiuc.edu><mailto:namd-l_at_ks.uiuc.edu<mailto:namd-l_at_ks.uiuc.edu>>" <namd-l_at_ks.uiuc.edu<mailto:namd-l_at_ks.uiuc.edu><mailto:namd-l_at_ks.uiuc.edu<mailto:namd-l_at_ks.uiuc.edu>>>
Subject: Re: namd-l: unreasonably high temp/pressure
Did you tell NAMD you are using TIP4P? you have to put "waterModel tip4" in your config file, the default is tip3.
~~Aron
On Wed, Oct 24, 2012 at 10:55 AM, Martin, Erik W <Erik.Martin_at_stjude.org<mailto:Erik.Martin_at_stjude.org><mailto:Erik.Martin_at_stjude.org<mailto:Erik.Martin_at_stjude.org>>> wrote:
I'm hoping someone on this list will have some insight on this problem I've been having. I'm running a simulation of an 82aa peptide in a truncated octahedron water box for a total of ~100k atoms. I'm also using an AMBER ff. I have been running this simulation for ~25ns using tip3 water with no problems at all. However, I wanted to run this with tip4 water as well and am seemingly incapable of getting it to equilibrate properly.
On the first structure, I was able to equilibrate it by constraining peptide coordinates and allowing the water – with rigid bonds – to equilibrate in the cell. When I rebuilt the system with tip4 water I did absolutely nothing different. It runs fine with rigid bonds on and the peptide constrained, but when I try to allow it to run I get the dreaded "atoms moving too fast" error at about step 120-200. It is different atoms every time, and what seems to be happening is that the temperature and pressure both ramp up to enormous values. (for the record, I'm using Langevin dynamics to control temp and pressure)
Does anyone have a clue what could be causing this condition?
Thanks a lot,
Erik
________________________________
Email Disclaimer: www.stjude.org/emaildisclaimer<http://www.stjude.org/emaildisclaimer><http://www.stjude.org/emaildisclaimer>
Consultation Disclaimer: www.stjude.org/consultationdisclaimer<http://www.stjude.org/consultationdisclaimer><http://www.stjude.org/consultationdisclaimer>
-- Aron Broom M.Sc PhD Student Department of Chemistry University of Waterloo -- Aron Broom M.Sc PhD Student Department of Chemistry University of Waterloo
This archive was generated by hypermail 2.1.6 : Mon Dec 31 2012 - 23:22:12 CST