


Next: xMDFF: MDFF for Low-Resolution
Up: MDFF Tutorial
Previous: MDFF with Domain Restraints
Contents
Subsections
MDFF with Symmetry Restraints
This section will show you how to set up a MDFF run using symmetry restraints.
Symmetry restraints use harmonic forces to maintain a symmetric structure during MDFF simulations for symmetric molecules.
The symmetric structure is determined by transforming and overlapping the atomic coordinates of all symmetric units and calculating the average positions of the transformed atoms.
Preparing the initial structure
For this example we will be using a nitrilase structure with helical symmetry, found in
helix.pdb.
The structure is already rigid-body docked into an experimental map (EMDB 1313) with Situs.
The map has been converted into the 3D potential for MDFF, named as helix-target.dx.
Please refer to previous section for use of Situs for rigid-body docking and use of mdff griddx for map conversion.
- 1
- Start a new VMD session.
- 2
- Load the initial structure in VMD by typing:
- 3
- Use the AutoPSF plugin as in
Section 2.1. If you are working on the same VMD session
from the beginning of the tutorial, make sure you click the Reset AutoPSF button and the choose the correct molecule in the AutoPSF plugin. Follow the same steps as before to make helix_autopsf.psf and
helix_autopsf.pdb.
- 4
- Generate a PDB file containing the per-atom scaling factors
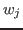 in Equation 1, as in previous section.
in Equation 1, as in previous section.
| mdff gridpdb -psf helix_autopsf.psf -pdb helix_autopsf.pdb |
|
| -o helix-grid.pdb |
|
- 5
- Generate secondary structure restraints as in previous section:
| package require ssrestraints |
|
| ssrestraints -psf helix_autopsf.psf -pdb helix_autopsf.pdb |
|
| -o helix-extrabonds.txt -hbonds |
|
- 6
- Generate restraints to prevent cis/trans peptide transitions
and chirality errors:
| mol new helix_autopsf.psf |
|
| mol addfile helix_autopsf.pdb |
|
| cispeptide restrain -o helix-extrabonds-cispeptide.txt |
|
| chirality restrain -o helix-extrabonds-chirality.txt |
|
We need to create a symmetry PDB file containing the designations of symmetric units, i.e. what the symmetric units are.
In the symmetry PDB file, the occupancy column denotes the "symmetry group"
atoms belong to, while the beta column denotes the symmetric units designation.
In this example, we have a single symmetry relationship, the helical symmetry, so we only have one symmetry group and hence the occupancy column is set to 1.
We have nine different symmetric unit within this helical symmetry group,
so the beta column of the first symmetric unit is set to 1 and
increases by 1 for the next symmetric until the last symmetric unit with beta column assigned to 9.
For more information on symmetry restraint parameters, please read the
documentation.
- 1
- Load the pdb file we are using for the initial structure
| mol new helix_autopsf.pdb |
|
- 2
- Assign beta and occupancy values for different symmetric units according to the rules described above. A tcl script has been provided to you for setting these values. Note that we are applying symmetric restraints to
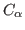 atoms only.
atoms only.
Next we have to create a matrix file which contains the transformation matrices needed to overlap the symmetric subunits based on their symmetry.
If the matrix file is not given to NAMD, NAMD will attempt
to generate these matrices automatically by guessing the symmetry information among the symmetric units.
This file follows a specific format outlined below:
- 1
- Matrices should be in order of symmetric units designation (beta column of the symmetry pdb file generated
above) e.g. The first matrix is applied to symmetric unit 1, the second matrix to symmetric unit 2 and so on.
- 2
- The matrices are defined as the transformation necessary to overlap the symmetric units
onto the first symmetric unit.
This means that the first matrix should be an identity matrix.
- 3
- The file should not have any leading or trailing blank lines.
Matrices should be separated by one and only one line.
The matrix itself should be a 4x4
transformation matrix with one row per line and each column separated by one space only.
For example, the identity matrix at the beginning of the file will look like the following:
1 0 0 0
0 1 0 0
0 0 1 0
0 0 0 1
Users can generate the 4x4 matrices for different transformations by using the measure fit command in VMD.
Please refer to the documentation
in the VMD user guide for more details.
The matrix file for the helical nitrilase system has been provided: helix-matrices.txt
NAMD configuration files for MDFF can be generated similarly to the previous example.
2.
| mdff setup -o symmetry -psf helix_autopsf.psf |
|
| -pdb helix_autopsf.pdb |
|
| -griddx helix-target.dx |
|
| -gridpdb helix-grid.pdb |
|
| -extrab {helix-extrabonds.txt
helix-extrabonds-cispeptide.txt |
|
| helix-extrabonds-chirality.txt} -gscale 1.0 -minsteps 2000 -numsteps 500000 |
|
- 1
- Now we need to edit the configuration file symmetry-step1.namd that we just
created by adding in the parameters necessary for symmetry restraints. Open
the file in any text editor and add the following lines anywhere before the source mdff_template.namd line:
| symmetryRestraints on |
|
| symmetryfile helix-symmetry.pdb |
|
| symmetryk 200 |
|
| symmetryMatrixFile helix-matrices.txt |
|
| symmetryfirststep 2001 |
|
| symmetryfirstfullstep 502000 |
|
These parameters turn symmetry restraints on and let NAMD know where to look for the
symmetry information.
The symmetryk entry is a constant which scales the harmonic force applied by the restraint. Thie value is scaled down by the number of atoms in a symmetric unit. Users can define a per-atom force constant which assign force constant to individual atoms instead of to the whole symmetric unit, which is discussed in the NAMD user guide.
One can vary the forces over time by modifying the force constant.
In this case, the force constant will be linearly increased over time, allowing more
conformational freedom at the beginning, working up to more rigid restraints as the
molecule is fitted to the density.
This is accomplished by the symmetryfirstfullstep entry which control when the force constant become the full assigned value (i.e. the symmetryk value).
Setting this value to last timestep will linearly increase
the force constant from the first timestep to the last timestep.
More information about these parameters can be found in the symmetry
restraint documentation
in the NAMD User's Guide.
- 2
- Quit VMD.
- 3
- Run NAMD using the configuration files generated by VMD, i.e.,
run the following commands in a terminal (or submit them to a cluster):
| namd2 symmetry-step1.namd > symmetry-step1.log |
|
This step should take about 8 hours on a modern quad core desktop.
If you don't want to wait, you can proceed to the analyzing section with the provided trajectory files.
The resulting trajectories will be saved to files symmetry-step1.dcd. If you want to continue working
through the tutorial before the simulations are complete, you can use the provided
trajectory files symmetry-step1-result.dcd instead.
First, you should load and view the trajectory file no-symmetry-step1-result.dcd provided.
This trajectory is the result of running the above simulation with symmetry restraints turned
off. You should notice that the last dimer is pulled
away from the molecule, seen in Fig. 12.
This is due to the extra density adjacent to these regions.
Figure 12:
MDFF simulation of nitrilase without symmetry
restraints. Red box indicates region of last dimer affected by adjacent
density.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/norestraint}
}
\end{center}
\end{figure}](img30.gif) |
While it is possible to
cut off extraneous portions of the map, this can introduce errors along the boundaries.
Instead, we can use symmetry restraints to avoid these distortions.
Load and view the trajectory obtained from your simulation
using symmetry restraints symmetry-step1.dcd or use the trajectory file provided
symmetry-step1-result.dcd.
Compare the movements of the first and last dimers to those without symmetry restraints.
Next: xMDFF: MDFF for Low-Resolution
Up: MDFF Tutorial
Previous: MDFF with Domain Restraints
Contents
school@ks.uiuc.edu