


Next: cis peptide bonds in
Up: Structure Check Tutorial
Previous: Introduction
Contents
Subsections
Chirality in proteins and nucleic acids
All amino acids but glycine have at least one chiral center at
C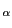 (see Fig. 1). Threonine and isoleucine have
an additional chiral center at C
(see Fig. 1). Threonine and isoleucine have
an additional chiral center at C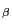 . According to the D-/L-
naming convention, naturally occurring amino acids are found in the
L-configuration. Note, however, that D-amino acids do occur in
biology, e.g., in cell walls of bacteria. Nucleic acids also have
chiral centers. For example, in DNA the atoms C1', C3', and C4' are
chiral, while RNA has an additional chiral center at C2'.
Chirality is central to all molecular interactions in biological
systems. A simple experiment demonstrates the principle: try to shake
someone's left hand with your right.
. According to the D-/L-
naming convention, naturally occurring amino acids are found in the
L-configuration. Note, however, that D-amino acids do occur in
biology, e.g., in cell walls of bacteria. Nucleic acids also have
chiral centers. For example, in DNA the atoms C1', C3', and C4' are
chiral, while RNA has an additional chiral center at C2'.
Chirality is central to all molecular interactions in biological
systems. A simple experiment demonstrates the principle: try to shake
someone's left hand with your right.
To demonstrate the usage of the plugin we will start by checking a
structure consisting of the protein EF-Tu in complex with a
tRNA
 . The used structure is based on the the PDB
structure 1OB2 in which errors have been introduced manually.
. The used structure is based on the the PDB
structure 1OB2 in which errors have been introduced manually.
Currently, the chirality plugin can be used from the text
console and through a GUI. The available commands provided by the
pugin can be obtained by typing chirality on the Tk
Console or the VMD console. The following information should be
printed in the console:
Commands:
check -- identify chirality errors
list -- list identified chirality errors
minimize -- fix chirality errors using energy minimization
move -- move hydrogen atom to fix chirality
reset -- reinitialize plugin state
restrain -- generate NAMD extrabonds file to prevent chirality changes
show -- visualize identified chirality errors
In the same way, the usage information for any of the provided
commands can be obtained. For example, typing chirality
restrain will show the syntax of the command that generates
restraints for the impropers at chiral centers which can be used in
NAMD.
In the following, we will describe how to use the chirality
plugin through its GUI, but all of the tasks can also be accomplished
from the console.
- 1
- Load the files chir_testcase.psf and
chir_testcase.pdb into a new session of VMD.
- 2
- Open the chirality GUI by selecting Extensions
 Modeling Fix Chirality
Errors in the VMD Main menu. In the upper part of the GUI
(see Fig. 2) the user can specify the molecule and
the atom selection to be tested.
Modeling Fix Chirality
Errors in the VMD Main menu. In the upper part of the GUI
(see Fig. 2) the user can specify the molecule and
the atom selection to be tested.
Figure 2:
The chirality GUI window.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.4]{FIGS/chirality_basic}
}
\end{center}
\end{figure}](img9.gif) |
- 3
- The button Check structure initializes the
test. Identified unusual chiral center configurations are displayed
in the Identified chirality errors form (see
Fig. 3). In the present case there are 9 chiral
errors identified in both the protein and RNA parts of the loaded
structure.
Figure 3:
Identified unusual chiral center
configurations in the chirality GUI window.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.4]{FIGS/chirality_errors}
}
\end{center}
\end{figure}](img10.gif) |
- 4
- By selecting an entry in the Identified chirality errors form
and hitting the button Show selected chiral centers it is
possible to visually inspect all the individual identified unusual
chiral centers. In the created representation (see
Fig. 4) the chiral center is highlighted by a
transparent purple sphere, while the four bonded atoms are shown in
CPK.
Figure 4:
Representation of the chiral error in U12
of the tRNA
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.4]{FIGS/chirality_show}
}
\end{center}
\end{figure}](img11.gif) |
- 1
- If the shown chiral center has the wrong chirality, the user can tag
an atom to be moved to flip the chirality at the selected center. This
is done by hitting the button hydrogen. Note, currently
only the moving of a hydrogen atom at a chiral center is supported. If
the chirality at the displayed center is correct, nothing needs to be
done. It is also possible to untag a tagged atom by selecting it and
hitting none.
- 2
- Once a chiral center has been inspected, and the atom to be moved
has been tagged, the actual moving of the atom can be executed by
hitting the button Move tagged atoms for selected chiral
centers.
- 3
- Since the described procedure (simple moving of the hydrogen atom)
generates an unphysical geometry of the molecule, it is necessary to
optimize the structure using an MD force field. The chirality
plugin uses the AutoIMD plugin for this purpose. This final
step is accomplished by selecting the chiral centers which should be
relaxed and hitting the button Minimize/equilibrate
selected chiral centers. This will open the AutoIMD
Controls window (see Fig. 5).
Figure 5:
AutoIMD window called by chirality plugin.
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.4]{FIGS/autoimd_basic}
}
\end{center}
\end{figure}](img12.gif) |
- 4
- Select Settings Minimization Mode in the
AutoIMD Controls window and hit then the Submit
button. Once the AutoIMD session started, hit the Connect button and a minimization will start. Finish the
simulation when the structure relaxed. Usually, it takes up to a few
thousand steps until atoms do not move anymore. Is the structure
minimized, hit the Finish in the AutoIMD Controls
window.
For more details about using the AutoIMD plugin, the user is
referred to the AutoIMD user's guide
http://www.ks.uiuc.edu/Research/vmd/plugins/autoimd/
.
- 5
- WARNING: Depending on the quality of the initial structure, it
can happen that not all errors can be fixed in one run. Thus, it is
important to check the final result again and correct the remaining
errors, if necessary. In order to check the structure after the
performed minimization/equilibration using AutoIMD, the chirality
plugin should be reset. This can be done by hitting the Reset chirality plugin button at the bottom of the
chirality window. Note, in the current implementation it will be
necessary to select the molecule again in the top part of the
chirality window. Note also that if the molecule to correct is
solvated, it may be necessary to equilibrate the minimized parts of
the structure. This can be done by selecting Settings Equilibration Mode and submitting a new AutoIMD run.
- 6
- Finally, it is a good advice to minimize the structure in the very
last step. This should be done by selecting Settings Minimization Mode in the AutoIMD
Controls window, which is still open from the last AutoIMD
session, and hit then the Submit button. As mentioned in
the introduction, additional restraints during the correction of
chirality errors. The restraints will be used always when AutoIMD is started from the chirality window. The
equilibrium values for these restraints are very approximate and
bring the configuration in the correct region. Thus, in order to
obtain a optimized structure, it is necessary NOT to use the
restraints defined by chirality. This is achieved by reusing
the AutoIMD window from the previous
minimization/equilibration; the restraints will not be taken into
account.
- 7
- Once all minimization/equilibration steps are performed, the corrected structure
should be saved. This can be done in the following way. Select the molecule in the VMD main window.
Create a representation containing all atoms in the Graphical Representations window.
Select File Save Coordinates in the VMD main window.
Select all in the Selected atoms and hit Save. Finally, specify the file
name in the Chose filename to save trajectory and hit OK.
As mentioned in the introduction,
structure optimizations and MD
protocols applying external forces can introduce chirality errors into
the simulated structure. To preserve stereochemistry, the chirality plugin offers the possibility to generate restraints
in form of extrabonds which can be used in a NAMD simulations.
This feature currently is available only from the console.
- 1
- Assuming the structure of interest is loaded in the top molecule in
VMD and one wishes to obtain restraints for all chiral centers in
proteins and nucleic acids, the command reads
chirality restrain -o chirality-extrabonds.txt
The file chirality-extrabonds.txt contains now two improper
restraints for each chiral center. One includes the chiral center and
its non-hydrogen binding partners. In the second restraint, the chiral
center is replaced by the hydrogen bound to it. The equilibrium values
of the two impropers are set to the values present in the used structure.
WARNING: Note, the specified restraints modify the potential
energy function of the system, and thus should not be used in
equilibrium production runs. Rather, these restraints are supposed to
be used only in simulations in which large forces occur due to poor
initial geometry or due to externally applied forces, e.g., MDFF or
TMD. Note also that although the final structure resulting from such
simulations will be correct, the behavior of the system during the
simulation is artificial.
Next: cis peptide bonds in
Up: Structure Check Tutorial
Previous: Introduction
Contents
school@ks.uiuc.edu
![\begin{figure}\begin{center}
\par
\par
\latex{
\includegraphics[scale=0.5]{FIGS/chirality}
}
\end{center}
\end{figure}](img6.gif)
{kind=link}