QwikMD Spotlights
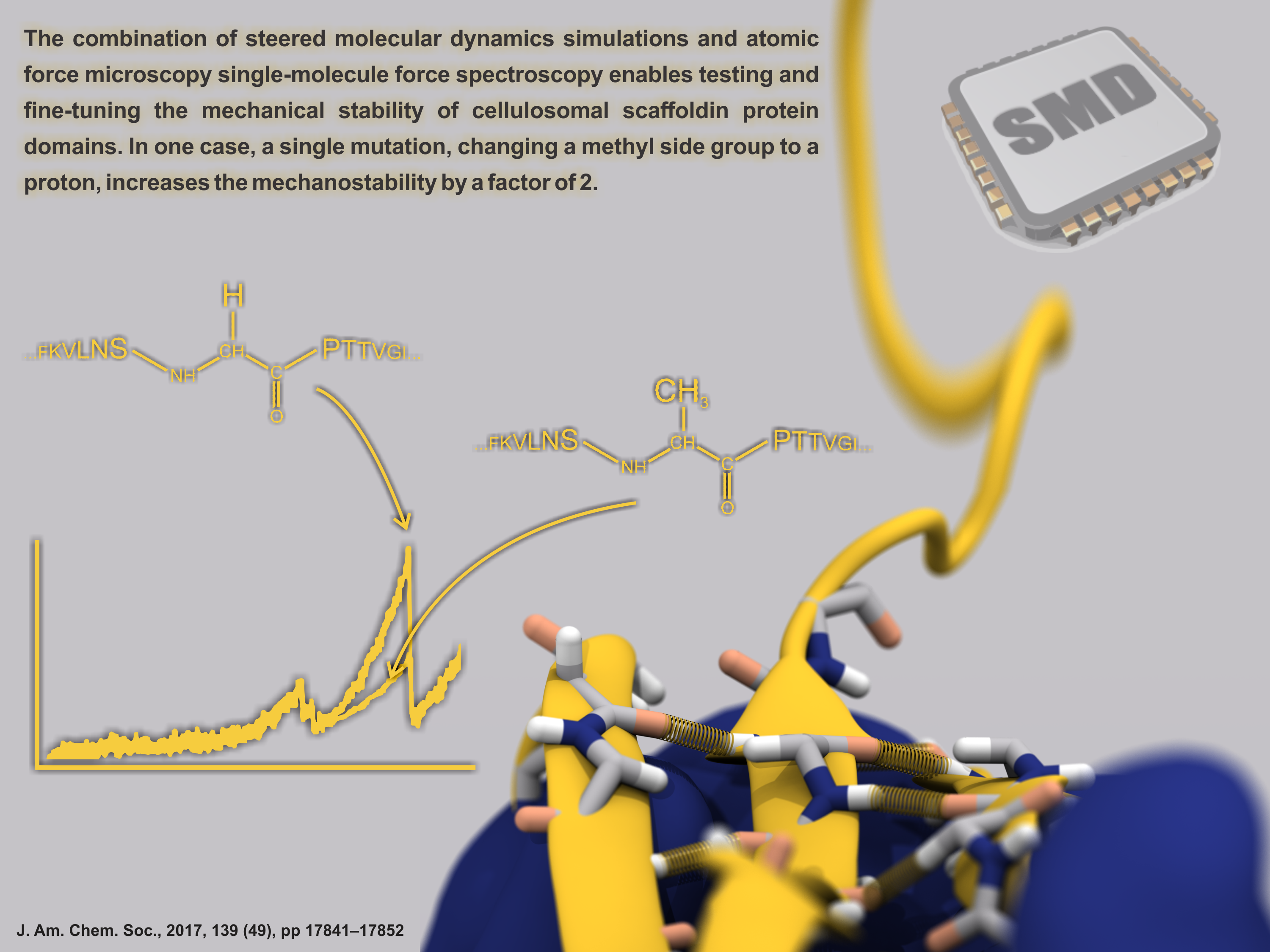
image size: 303.2KB
Proteins play major mechanical roles in a cell. Mechanical properties of proteins can be substantially modulated by slight changes in their amino acid composition, an ability essential to bacterial cells, which use protein repeats with small modifications to tune their mechanical strength, and thus their functional properties. A prime example of the role of biomechanics in the cell is found in symbiont gut bacteria who need to hold to their food source in such turbulent environments as the rumen of the cow. Proteins evolved for this purpose are known actually to be among the strongest mechanical biomolecules. Key to the process are molecular tentacles, so-called cellulosomes, on the surface of symbiotic bacteria. The cellulosomes develop a tight grasp on and then effective cleavage of hardy cellulose fibers of the grass. In a joint experimental-computational study researchers investigated the mechanical properties of cohesin, a major cellulosome component, under force. Using NAMD, all-atom steered molecular dynamics (SMD) simulations on homology models offered insight into the process of cohesin unfolding under force. Based on the differences among the individual force propagation pathways and their associated correlation communities, the researchers were capable of designing protein mutants to tune the mechanical stability of the weakest cohesin. The proposed mutants were tested with high-throughput atomic force microscopy experiment revealing that in one case a single alanine to glycine point mutation suffices to more than double the mechanical stability. Read more about our cellulosome research here.
image size:
507.7KB
made with VMD
The 2.12 release of the molecular dynamics program NAMD provides major enhancements in performance, flexibility, and accuracy, complementing the greatly enhanced usability provided by the QwikMD GUI released in VMD 1.9.3. NVIDIA GPU-accelerated simulations with NAMD 2.12 are up to three times as fast as 2.11, particularly for implicit solvent simulations and single-node simulations of smaller systems. NAMD 2.12 is also optimized for the new Intel Xeon Phi KNL processors found in Argonne Theta, NERSC Cori, and TACC Stampede 2. NAMD 2.12 builds on the asynchronous multi-copy scripting capabilities introduced in NAMD 2.11 with the ability to modify and reload the molecular structure, enabling development of grand canonical and constant pH ensemble methods, as well as an optional Python interface for advanced on-the-fly analysis. Finally, NAMD 2.12 provides a complete, no-recompilation-needed interface for hybrid QM/MM with both the semi-empirical code MOPAC and the ab initio/DFT code ORCA. More on new features in the 2.12 release of NAMD can be found here. NAMD is available free-of-charge as source code, precompiled binaries, pre-installed at supercomputer centers, and now jointly with VMD as one-click interactive molecular modeling on the Amazon cloud.

image size:
2.2MB
made with VMD
The latest release of VMD brings many advances that help researchers prepare, analyze, and visualize molecular simulations. The new QwikMD plugin streamlines key simulation preparation and analysis tasks, and guides users in the creation of reusable simulation workflows and protocols. VMD now includes several advanced features for parallel analysis and visualization of cellular-scale simulations, as reported here, and here. VMD 1.9.3 strengthens collaboration between experimental and computational biologists by supporting a broader range of experimental density map image formats, such as those used in cryo-electron tomography. Many updated plugins are included in VMD 1.9.3, including tools for analysis of free energy perturbation simulations, MDFF hybrid structure fitting, ffTK force field parameterization, and normal mode analysis. VMD 1.9.3 adds support for new hardware and operating system platforms including IBM OpenPOWER (ORNL Summit), a variety of GPU-accelerated ARM SoCs, the Amazon AWS EC2 cloud, and most recently, the Intel Xeon Phi Knight's Landing many-core CPU (TACC Stampede 2, Argonne Theta). The VMD 1.9.3 release adds stunning graphics produced using interactive ray tracing using the latest multi-core CPUs and GPU accelerators, enabling 360-degree panoramic movie rendering for VR headsets, as reported here, and here. Interactive ray tracing makes the task of getting a molecular image "just right" much easier than ever before; it also enables rendering of spectacular movies for communication of scientific results. A VR movie rendering tutorial assists users with the steps required in rendering and encoding VR movies for upload to YouTube for display using VR headsets such as Google Cardboard, Oculus Rift, and GearVR. More details about VMD 1.9.3 features can be found here.

image size:
1.2MB
made with VMD
Everything that living things do can be understood in terms of jigglings and wigglings of atoms.
Richard Feynman's remark in the early 1960's summarizes what is today widely accepted, namely,
that molecular processes can be described by the dynamics of biological molecules, therefore connecting
protein dynamics to biological function. Molecular dynamics (MD) is by far the best tool to investigate
jigglings
and wigglings
of biological systems. Advances in both software and hardware
have spread the use of MD, however the steepness of the learning curve of the methodology of MD
remains high. To assist new users in overcoming the initial barrier to use MD software, and to help the more advanced users to
speed up tedious steps, we have developed the QwikMD software, as decribed in a recent paper.
By incorporating an easy-to-use point-and-click user interface that connects the widely used molecular graphics
program VMD
with the powerful MD program NAMD,
QwikMD allows its users to prepare both basic and advanced MD simulations in just a few minutes. At the same time,
QwikMD keeps track of every step performed during the preparation of the simulation, allowing easy reproducibility
and shareability of protocols. More information about QwikMD, as well as introductory tutorials are available on our
QwikMD webpage.
QwikMD is available in VMD 1.9.3 or later versions.

image size:
299.6KB
While waste recycling in daily life has become popular only recently, living cells have been recycling their protein content since the very beginning. Recycling of unneeded protein molecules in cells is performed by a molecular machine called the proteasome, which cuts these proteins into smaller pieces for reuse as building blocks for new proteins. Proteins that need to be recycled are labeled by tags made of poly-ubiquitin protein chains. The proteasome machine recognizes and binds to these tags, pulls the tagged protein close, then unwinds it, and finally cuts it into pieces. Despite its substantial role in the cell's life cycle, the proteasome's atomic structure and function still remain elusive. In our recent study, we obtained an atomic structure of the human 26S proteasome by combining computational modeling techniques, through molecular dynamics flexible fitting (MDFF) of the cryo-electron microscopy (cryo-EM) data. The features observed in the resulting structure are important for coordinating the proteasomal subunits during protein recycling. One of the key advances is that for the first time the nucleotides bound to the ATPase motor of the proteasome are resolved. The atomic resolution of the structure permits to perform molecular dynamics simulations to investigate the detailed proteasomal function, in particular the protein unwinding process of the ATPase motor. Furthermore, our obtained structure will serve as a starting point for structure-guided drug discovery, developing the proteasome as a crucial drug target. The atomic models are deposited in the protein data bank (PDB) with the PDB IDs 5L4G and 5L4K and the 3.9 Å resolution cryo-EM density is deposited in the electron microscopy data bank EMD-4002. More information about our proteasome projects is available on our proteasome website. Easy access to our modeling techniques is provided through QwikMD, which was employed here for the first time.


