Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001

image size:
77.5KB
made with VMD
Did you get your flu shot this year? Influenza is a leading cause of preventable death in the industrialized world, representing hundreds of billions of dollars in healthcare expenditures and loss of economic production. While the yearly influenza vaccination is nearly 90% effective at limiting infections in populations less than 65 years of age, there is insufficient evidence regarding the effectiveness of the flu shot for the elderly population, whose immune systems may not mount an adequate antibody response to vaccination. Beyond vaccination, front-line therapies such as the neuraminidase inhibitors Tamiflu and Relenza have proven to be of limited effectiveness due to the evolution of drug-resistant influenza mutants. Therefore, a need exists for the development of new therapies to circumvent these resistance mechanisms. Computational biologists employing NAMD and VMD used molecular simulations to uncover the key role that water plays in mediating how well antiviral drugs can bind to proteins of the influenza virus. This investigation, reported recently, reveals that amino-acid mutations responsible for drug resistance act by reshaping the local electric field and also by permitting infiltration of water within the otherwise hydrophobic drug binding pocket. These mutations thus induce drug resistance in much the same way as inverting the polarity of a magnet can repel rather than attract. These findings are expected to help guide the design of novel drugs with increased antiviral efficacy. Additional details about this study can be found here.

All living systems contain proteins whose job is to move ions across a lipid membrane. Even viruses encode ion transport proteins, which they need to complete their lifecycle and release themselves from infected cells. Such proteins, called viroporins, usually consist of small subunits of one or two helices that can self-assemble in a lipid bilayer into a pore-like structure. Although in some cases, the resulting structures resemble the well-ordered, selective ion channels in higher organisms, often they take on a more disordered character, forming pores with variable numbers of subunits, which adapt their structure and behavior to the environment in which they find themselves. This inherent flexibility and disorder makes it very difficult to produce high-resolution crystal structures of viroporins, which is unfortunate, since they could offer attractive drug targets for new antiviral therapies. Computational modelling and molecular dynamics simulations can help fill in the gaps in our structural knowledge of viroporins, and provide plausible 3-D models for visualization and drug design. In a recent publication, scientists published models of the p7 viroporin found in Hepatitis C virus. MD simulations of these models revealed that p7 can form stable pores with 4 to 7 subunits, with a bias towards 6 or 7 subunits, and that the p7 oligomers are highly flexible in adapting to different membrane thicknesses. These simulations also suggested that specific amino acids in certain places in the structure could play a role in controlling the ion permeability of p7.
More details can be found on our p7 website.

Nanoengineering permits the manufacturing of sensors of unprecedented accuracy to detect biomolecules at very low concentrations as they arise, for example, as signals in living cells. In an important type of cellular signaling, proteins are modified through addition of a phosphate group by other proteins, so-called kinases. Kinases are involved in various types of cancers; therefore, bioengineers seek to develop a nanosensor to detect kinase activity. As described in a recent report, they grafted short peptides, containing a tyrosine amino acid, on nanometer scale gold surfaces. Phosphate groups are negatively charged, and as the groups are transferred from the kinases to the peptides tyrosine, the overall charge of the grafted peptides increase. Bioengineers detected then the phosphorylated peptides by applying electrical fields that would drive the charged phosphate group towards the surface or away from it, depending on the voltage polarity; the resulting conformational change of peptides can be recognized by shining light on the nanosensors as optical properties of molecules near metal surfaces are amplified. In order to make the nanodevice really work, the bioengineers needed to optimize the peptide sequence, know how phosphorylation and voltages alter the near-surface conformation of the peptides and how to interpret the measured optical signals. In other words, they needed a microscopic view of the nanodevice! Such view was achieved through molecular dynamics simulations using NAMD and VMD following in the footsteps of similar earlier uses of such simulations as a computational microscope (see Diet and DNA, Sep 2011; Bumpy DNA, Feb 2009; Stretchable DNA, Nov 2005). The combination of nanoengineering and molecular dynamics simulations produced indeed a satisfactory kinase sensor prototype. For more information, visit our kinase sensor website.

image size:
76.8KB
made with VMD
The fall flu season is coming. It is time to get your flu shot! Many people may still remember the influenza A H1N1 flu ("swine flu") pandemic of 2009, which caused 280,000 deaths worldwide. The best way to prevent the flu is to get vaccinated with a flu shot or use the flu nasal spray vaccine. However, rapid evolution of the flu virus constantly requires new vaccines. Fortunately, the immune system has several defensive mechanisms in the lung to clear inhaled pathogens. One of these mechanisms involves surfactant proteins which induce aggregation of viral particles and, thereby, prevent infection, serving as a front-line host defense. Recently, researchers found that surfactant protein D (SP-D) from pigs exhibits particularly strong inhibitory activity, more so that human SP-D. This discovery leads researchers to investigate SP-D structure-related antiviral activity. In a recent experimental-simulation study, crystallographic analysis of pig and human SP-D showed that a loop involved in viral binding on pig SP-D is longer than the respective loop on human SP-D; molecular dynamics simulation revealed that the longer loop of pig SP-D has higher flexibility than that of human SP-D, suggesting that the flexible loop region could facilitate strong binding of SP-D to virus. Based on this finding, one can develop new nasal spray anti-flu protection through other structural modification of human lung surfactant proteins. More on our lung surfactant protein website.

Hearing, sight, touch, taste, and smell are the five basic senses that link animals and humans to their habitat. In particular,
smell, or olfaction, endows animals and people with the ability to detect and distinguish different scents through volatile
odorant compounds and, thus, provides a crucial ability to recognize food or evade predators. The five senses have been studied
extensively and are believed to be well characterized, but remarkably the fundamental mechanism of olfaction is still debated.
The mainstream explanation of smell is based on recognition of the odorant molecules through characteristics of their surface,
e.g., shape, but certain experiments suggest that such recognition is complemented by recognition of vibrational modes.
As recently reported,
according to the latter suggestion, an olfactory receptor is activated by electron transfer assisted
through odorant vibrational excitation. The hundreds to thousands of different olfactory receptors in an animal recognize
odorants over a discriminant landscape with surface properties and vibrational frequencies as the two major dimensions.
The analysis revealed a range of physical characteristics which olfactory receptors and odorants must obey for the
vibrationally assisted electron transfer mechanism to function. More details are provided on our
olfaction website.
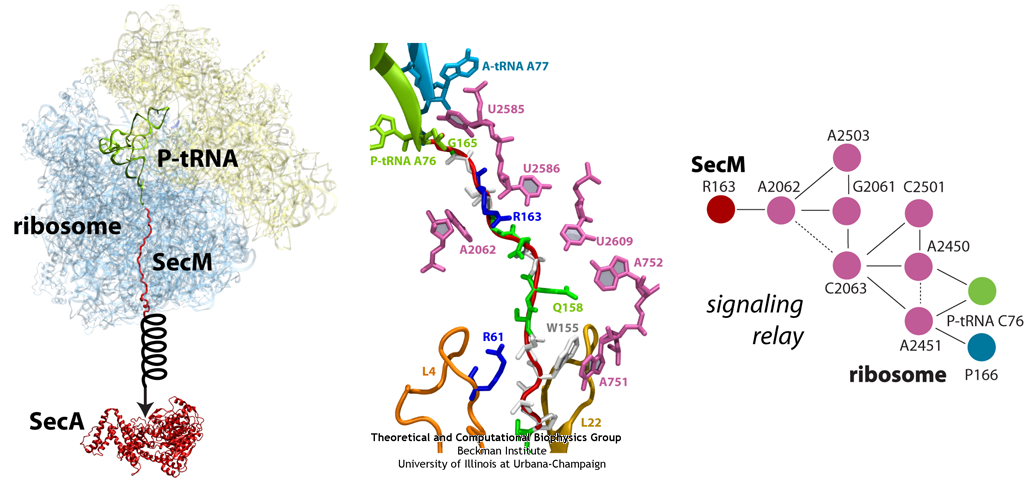
image size:
299.9KB
made with VMD
The ribosome functions as a cellular protein factory, synthesizing practically all the proteins in the cell based on blueprints read from DNA (see the April 2012 highlight and Dec. 2009 highlight).
However, unlike an assembly line, the ribosome has no foreman directing it. Instead, regulation of protein synthesis is managed by a number of external, and
internal, signals. For example, the protein TnaC halts its own synthesis in the ribosome to promote that of another protein (see the May 2010 highlight). Similarly, synthesis of the protein SecA, a translocase that aids in pushing newly
made proteins across membranes, is controlled through the nascent protein SecM. Regulation of SecA levels is the only function of SecM, which is degraded as
soon as it leaves the ribosome. It is the stalling of one ribosome by SecM that provides enough time for secA, which resides on the same messenger RNA
as secM, to be translated by a second ribosome, thus upregulating SecA production. When enough SecA has been produced, it pulls on the portion of SecM
outside the ribosome, relieving its stalled state. The critical interactions that cause stalling have now been identified through a combination of molecular
dynamics and cryo-electron microscopy via MDFF and NAMD. As recently reported, these interactions form a relay connecting SecM in the exit tunnel to the
ribosome's key catalytic center, preventing synthesis and thus explaining how SecM stalls inside "its" ribosome. Additionally, steered MD simulations revealed
how SecA can cause the nascent SecM to become unstuck, by breaking those same interactions. More details are provided on our ribosome website.

Proteins are the biological workhorses in living cells. For example,
they respond to external signals arriving at the cell surface or transport cargo,
much larger than themselves, from one place to another in the cell.
However, before a protein can carry out his job, it must first assume the proper shape.
Proteins are long polymers of twenty different amino acids linked in a linear sequence;
the latter is particular for each protein.
It is still a mystery how a protein folds into the proper shape based on its sequence.
Scientists hope that one day they can "watch" this folding process for any given protein.
The dream has been realized, at least partially, through the use of computer simulation.
After tackling the protein-folding problem already computationally for two small proteins
(see May 2008 highlight and
Nov 2009 highlight),
researchers have now successfully visualized the complete folding process of a relatively large protein,
the so-called λ-repessor
(see movie, 7.8 MB).
In fact, it is one of the largest proteins folded to date using a computer. As reported recently,
simulations carried out with the program NAMD,
as well as simulations carried out on a special purpose supercomputer, Anton,
achieved to follow λ-repessor's folding movement for more than 0.0001 seconds,
long enough to observe the protein assume its proper shape.
More information is available on our protein folding website.
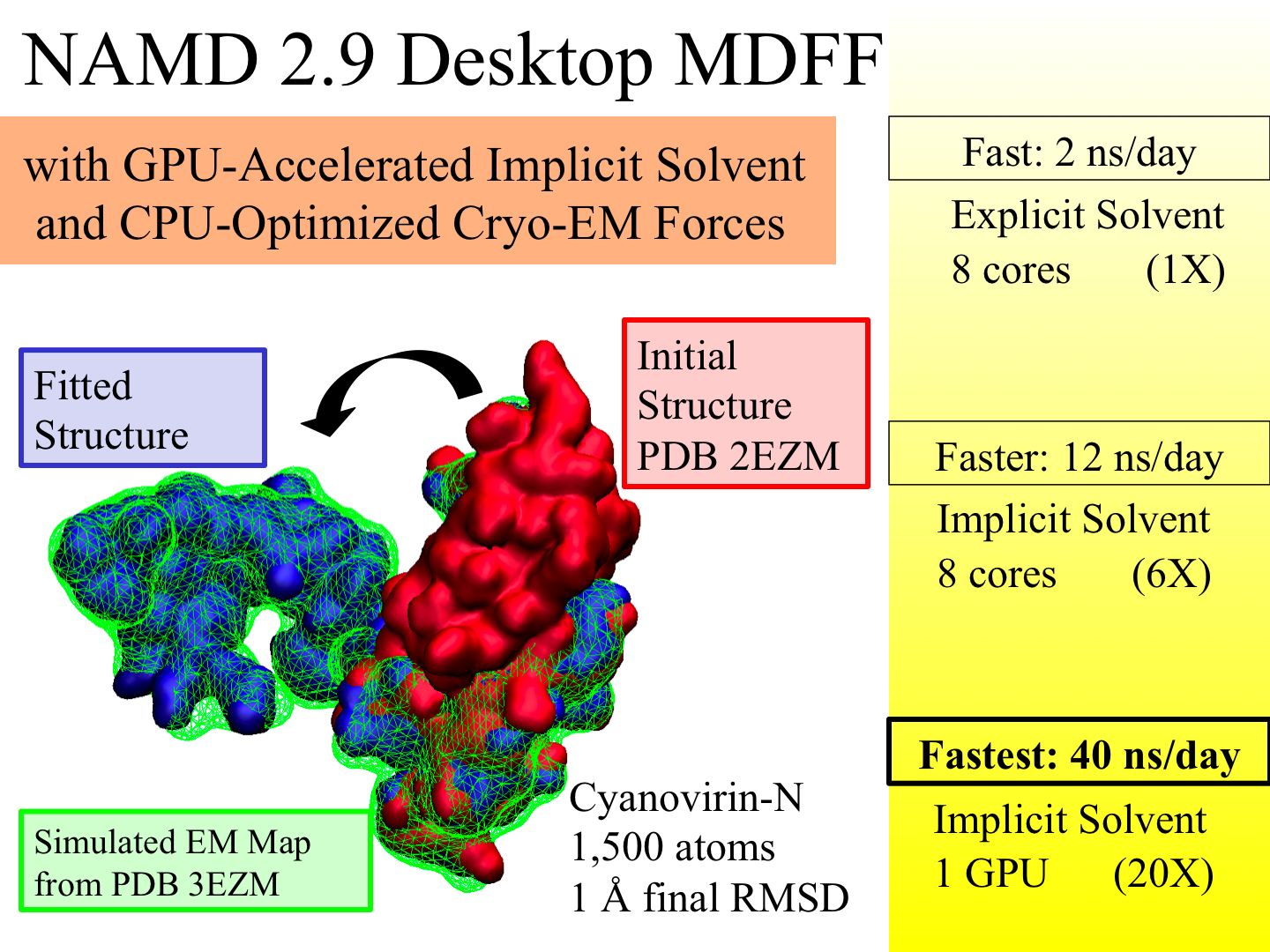
image size:
883.7KB
made with VMD
X-ray crystallography resolves the structures of the
molecular machines in living cells at an atomic level of
detail, but only in states that can be captured as
crystals, which are often not functional states.
Cryo-electron microscopy enables a more complete view of
biomolecular conformational variability, but at lower resolution.
The molecular dynamics flexible fitting (MDFF)
method combines the atomic detail of crystallographic
structures with lower-resolution cryo-electron microscopy
to synthesize all-atom models of complex macromolecular
aggregates such as the
ribosome in multiple functional
states. The 2.9 release
of NAMD combines
GPU acceleration
of implicit solvent
simulation,
optimizations exploiting shared memory within a single
machine, and a faster "lite" grid forces implementation
to bring MDFF capability from the supercomputer to the
desktop.
VMD can connect to a running simulation
to visually monitor the progress of the simulation or
to interactively steer a molecule
with either the mouse or a haptic (force-feedback) interface device.
The convergence of methodology, software, and hardware
advances thus opens what was once the domain of extremely
expensive equipment to commodity computers.

made with VMD
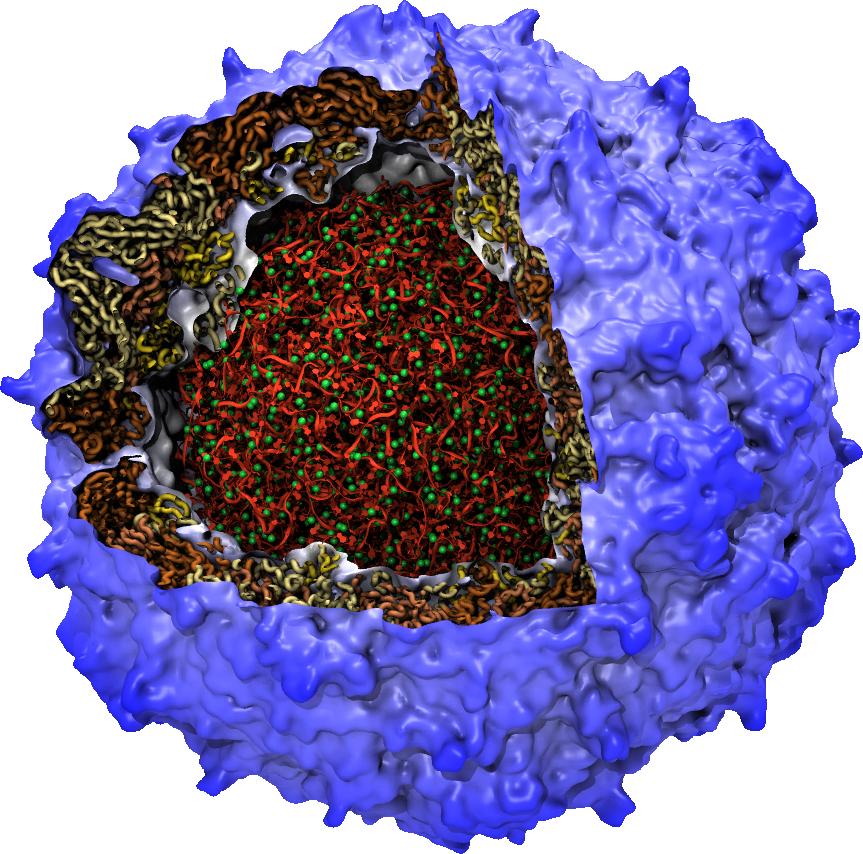
Viruses reproduce by splicing their genetic material into a host cell,
causing the cell to manufacture new viruses.
This genetic material is protected outside of the host cell by a protein
capsid, which disassembles inside a new cell to complete the infection process.
Simulation of viral infection
has progressed significantly since the
first all-atom virus simulation
was done with NAMD in 2006 and is one of the
driving biomedical projects for the software.
A new collaboration with the
Pittsburgh Center for HIV Protein Interactions
has applied molecular dynamics flexible fitting
to construct the first all-atom structure of an HIV virus capsid in its
tubular form (shown).
This structure is now being simulated as one of six
early science projects
on the
Blue Waters petascale supercomputer
being installed at Illinois.
These large-scale simulations are enabled by the
2.9 release
of NAMD, which includes a new high-performance
interface to the Cray Gemini network of Blue Waters.
Smaller simulations may also leverage petascale computing through a new
replica-exchange framework that supports parallel tempering and integrates
with the collective variables module for umbrella sampling conformational
free energy calculations.
GPU acceleration
enhancements include minimization and implicit solvent support as well
as exploitation of shared memory, extending performance gains to the desktop.
The surfaces of biomolecules are alive with activity, with surface shape
and electrostatic interactions leading them to interact with each other.
The recent
VMD 1.9.1 release
includes a new "QuickSurf" graphical
representation for molecular surfaces that allows the dynamics of large
biomolecules to be animated interactively for the first time. VMD 1.9.1
even enables surface representations for many-million atom complexes.
QuickSurf uses fast algorithms, GPU computing techniques, and multi-core
CPUs to achieve astonishing performance. The algorithms behind QuickSurf
have been
recently reported.
VMD 1.9.1 adds many other new features
including a new
Force Field Toolkit
(FFTK) plugin that assists researchers
in development of CHARMM force field parameters, a new
NetworkView plugin
for mapping and displaying networks on 3-D molecular structures, an
updated
ViewChangeRender
plugin for making sophisticated demonstrations
and movies, and a new
VMD remote control
tool that allows VMD sessions to
be controlled from wireless touch sensitive phones and tablet devices.
For more on these and other new features of VMD see the
VMD 1.9.1 release page.

image size:
488.4KB
made with VMD
The ribosome is the protein assembly line in all living cells. The building material
for new proteins is supplied by RNA molecules, called tRNA. They
enter and move through the ribosome, each adding a new amino acid to the nascent proteins
according to the genetic sequence provided through so-called messenger RNA.
During the tRNAs' translation through the ribosome, the ribosome itself is not static either.
A ratcheting motion and other large scale motions can be observed. However, the exact
tRNA and ribosome motion were not clear.
Using images from cryo-electron microscopy,
MDFF,
a computational method based on NAMD,
allows one to see the moving parts within the ribosome in great
detail.
MDFF (see the June 2008 highlight) already provided crucial and unique insights into different
aspects of protein synthesis, such as
translational
arrest of the ribosome by a nascent chain or
translocation of
an emergent protein across a membrane.
In the work
reported recently,
MDFF revealed the presence of previously unseen intermediate states of the ribosome and its bound tRNAs during the ratcheting motion.
A thorough analysis of these states pictures the ribosome as a molecular machine using Brownian motion
for its function. More on our ribosome website.
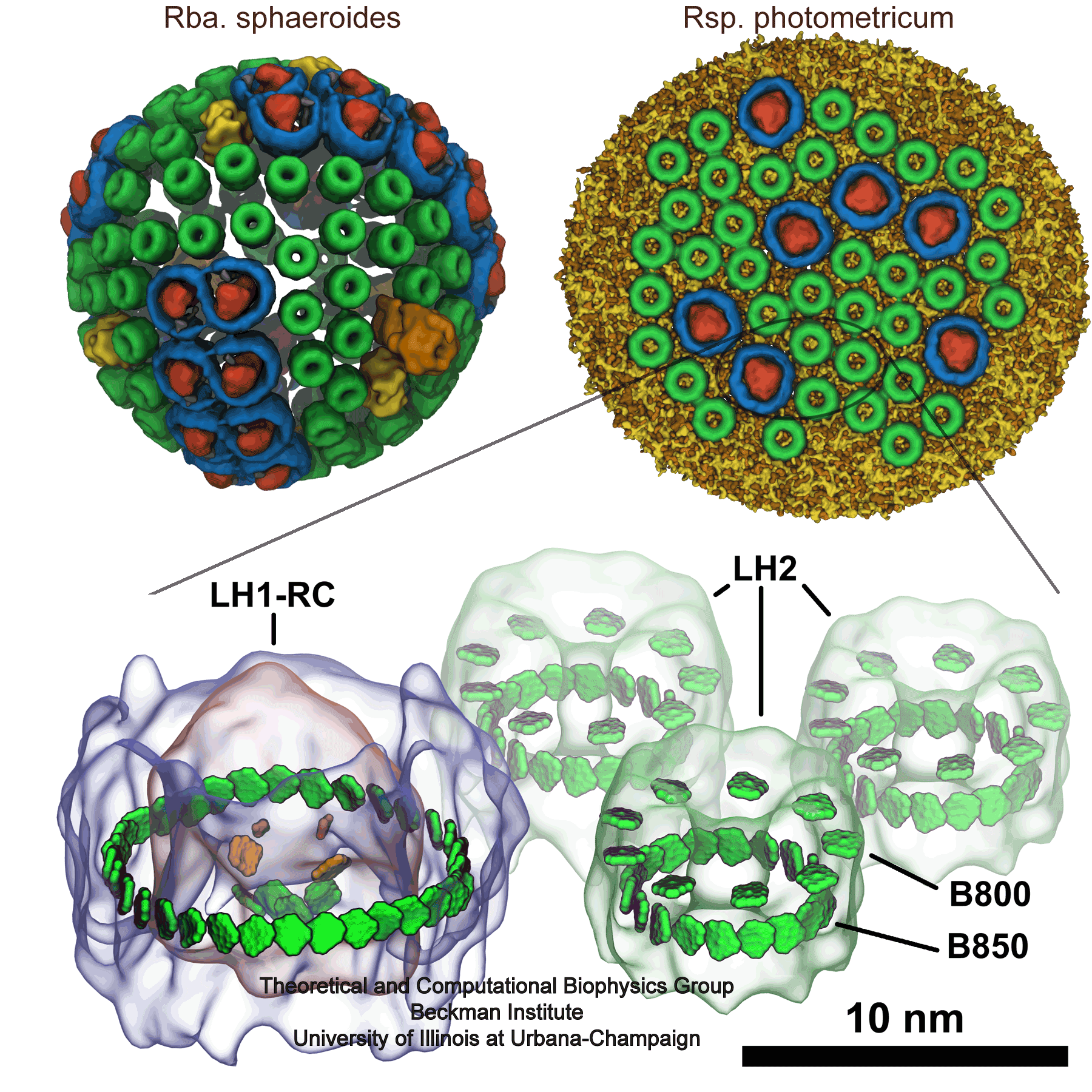
image size:
740.7KB
made with VMD
Many bacteria use sunlight as an energy source.
The energy gained from a solar ray absorbed by a molecule, however,
lasts only for a
very short time (a mere 0.000000001 seconds!) before it dissipates away
and is lost. Within this short time, machinery in the bacterial cell must
store the light energy in a longer-lasting form so that it can be used
later. A series of reviews describes how bacteria exploit quantum physics
to bottle the energy of sunlight for a sufficiently long time to fully
utilize it. In a first review
we introduce the light harvesting systems of bacteria and their key
molecular components, in particular the role of chlorophylls. In a second review we
describe how thousands of chlorophylls cooperate to transport the short
lived energy of absorbed light to the centers where the energy is converted into a more stable
form, namely that of a voltage difference across the bacterial cell
wall. In a
third review we explain how quantum physics enhances this process
of energy transport in bacteria (see also our video).
In a
fourth review we describe how the individual components of
this system come together into their overall organization.
More information
about the machinery and process of photosynthesis can be found here and about the physics of energy
transfer in photosynthesis here.
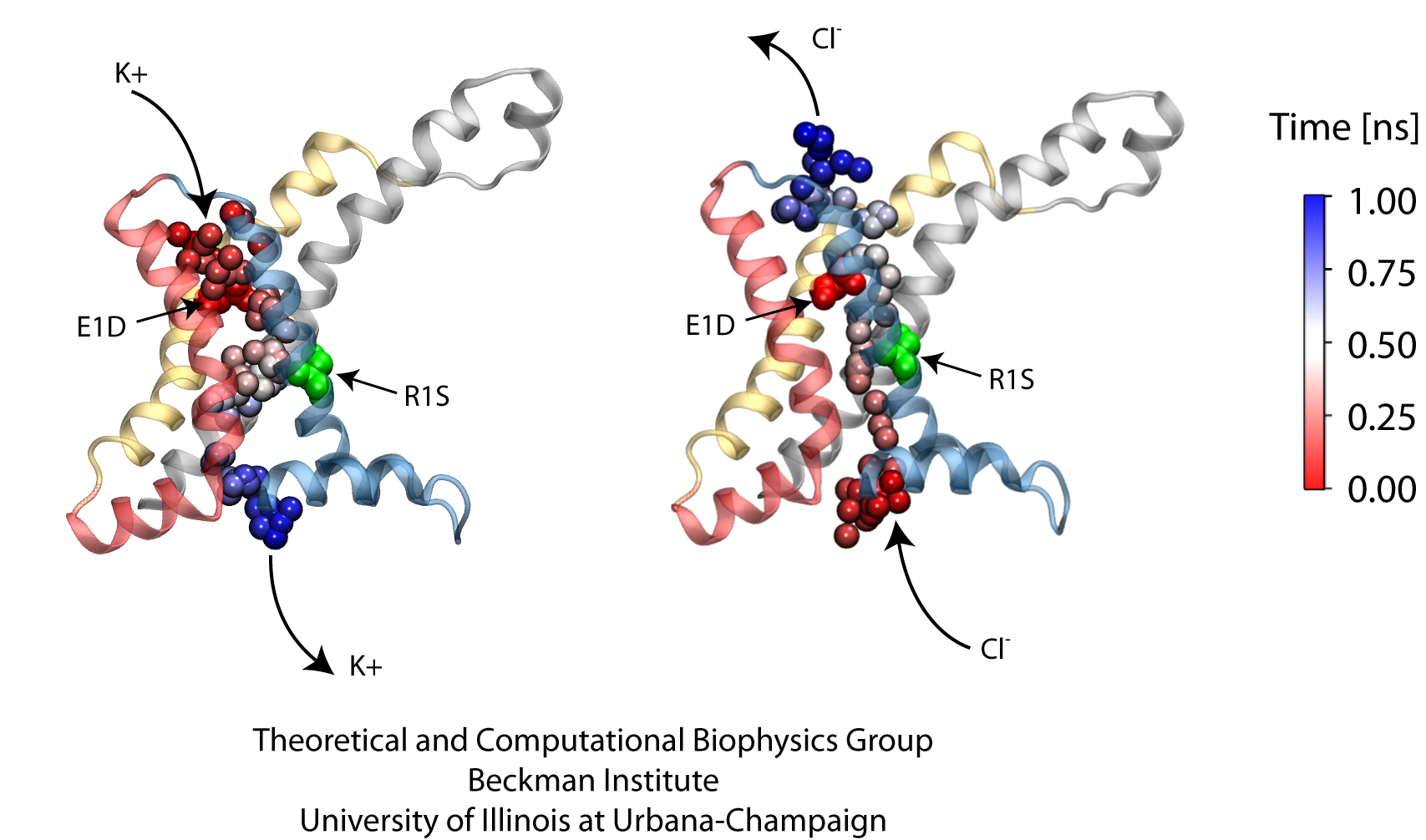
image size:
419.0KB
made with VMD
Voltage-gated ion channels, present in the membrane of excitable cells, control the ionic concentrations of the cellular environment by maintaining a potential difference of -100 mV between inside and outside of the cell membrane. Voltage-sensing occurs through distinct protein modules, known as voltage-sensor domains, four of which surround the main conduction pathway in potassium channels. Mutation of a certain amino acid on the voltage sensor domain turns these protein modules into cation channels, known as omega pores, which allow conduction of ions only when the main pathway is closed. Omega pores closely resemble the long-sought voltage-gated proton channels, which were recently identified to follow the same voltage-sensing mechanism as voltage-gated cation channels. In a recent report, researchers have visualized the twisted permeation pathway of the ions through omega pores using the molecular dynamics program NAMD. The simulations revealed a narrow constriction region lined by negatively charged amino acids, acting as a selectivity filter that prefers passage of positively charged ions through the pore.
For more detail, see our potassium channel website .

image size:
35.6KB
Courtesy of Vita Solovyeva
Creatures as varied as mammals, fishes, insects, reptiles, and birds have an intriguing 'sixth' sense that allows
them to navigate in the Earth's magnetic field. Despite decades of study, the physical basis of this sense remains
elusive. A likely mechanism is furnished by magnetic field sensitive reactions occurring in the retina of animal eyes.
A decade ago it was suggested (see our magnetoreception page)
that the photoreceptor cryptochrome, arising in the retina, endows birds with magnetoreceptive abilities. The
hypothesis gained support during the last years, after it had been shown that the protein exhibits properties
required for an animal magnetoreceptor to operate properly.
(see prior highlights on
A Compass in the Eye, July 2010;
on
Where's North, Ask Superoxide, July 2009;
and on
Animal Magnetic Sense Shared by Plant, April 2007).
However, the biophysical mechanism of cryptochrome
activation and signaling is still poorly understood.
A recent study
proposed a theoretical analysis method for
identifying cryptochrome's signaling reactions involving comparison of measured and calculated reaction kinetics.
Application of the method suggest a light-driven reaction cycle which combines electronic excitation with
electron and proton transfer reactions in the protein. More details on cryptochrome functioning as a
light-driven magnetic compass can be found on our
cryptochrome webpage .
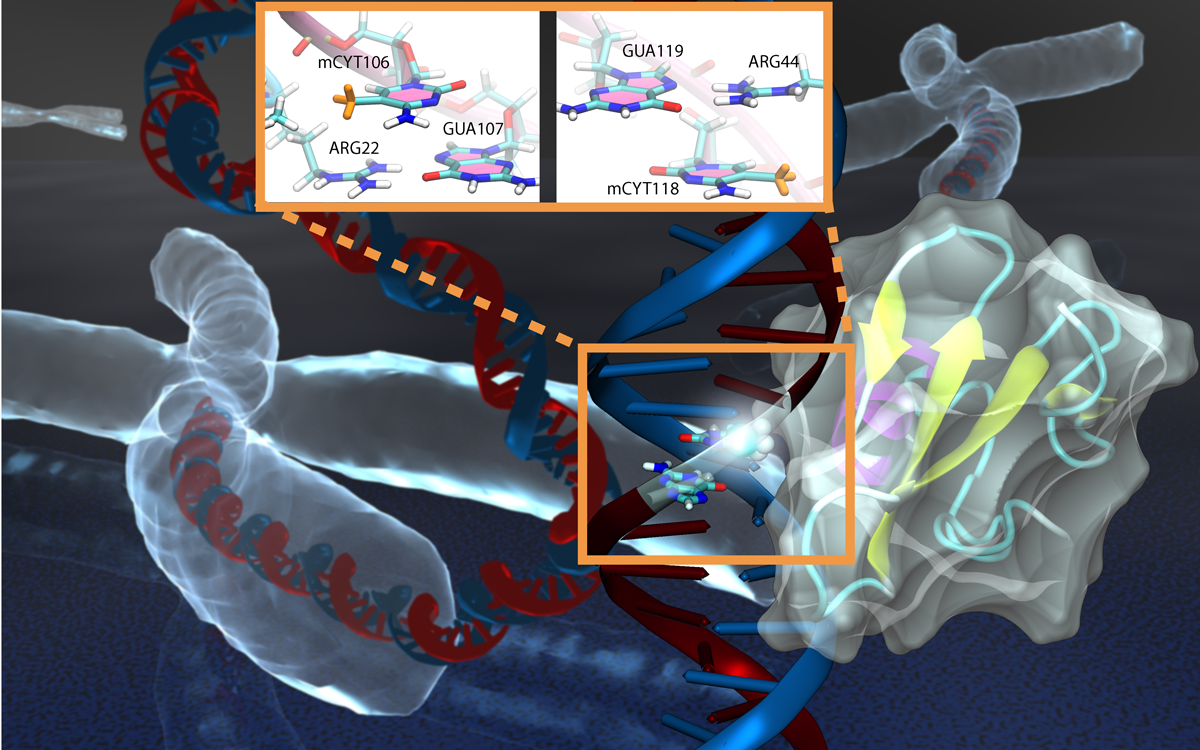
image size:
85.6KB
made with VMD
Genes encoded in DNA sequence give a complete set of instructions for the development of a new organism. However, an organism, like the human body, develops also over a life time adapting to environment and experience, for example to diet and exercise. Recently, researchers found that such factors act through so-called epigenetic mechanisms that alter an organism's development without altering DNA sequence. One such mechanism involves DNA methylation, a chemical modification of one of the four bases of DNA, cytosine, that replaces a hydrogen atom with a methyl group. There are several ways that DNA methylation exerts its biological function, bringing about a long-time adaptation of an organism to its environment, in some cases even across generations. Our previous experimental and computational studies (see Sep 2011 and Feb 2009 highlights) indicated that methylation changes mechanical properties of DNA which can affect gene expression. DNA methylation can also inhibit gene expression by impeding proteins that control the translation of DNA sequence into protein synthesis. One mechanism involves DNA methylation sites recruiting genetic control proteins that inhibit DNA expression through their local presence. In a recent study, computational biologists performed MD simulations with NAMD along with quantum chemistry calculations to determine recognition of methylated DNA by proteins. The simulations revealed how a certain genetic control protein, called methyl-CpG binding domain protein, acts in tandem with methylated DNA like a key and a lock, methylated DNA and protein perfectly matching each other. More details can be found on our methylated DNA website.


