Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
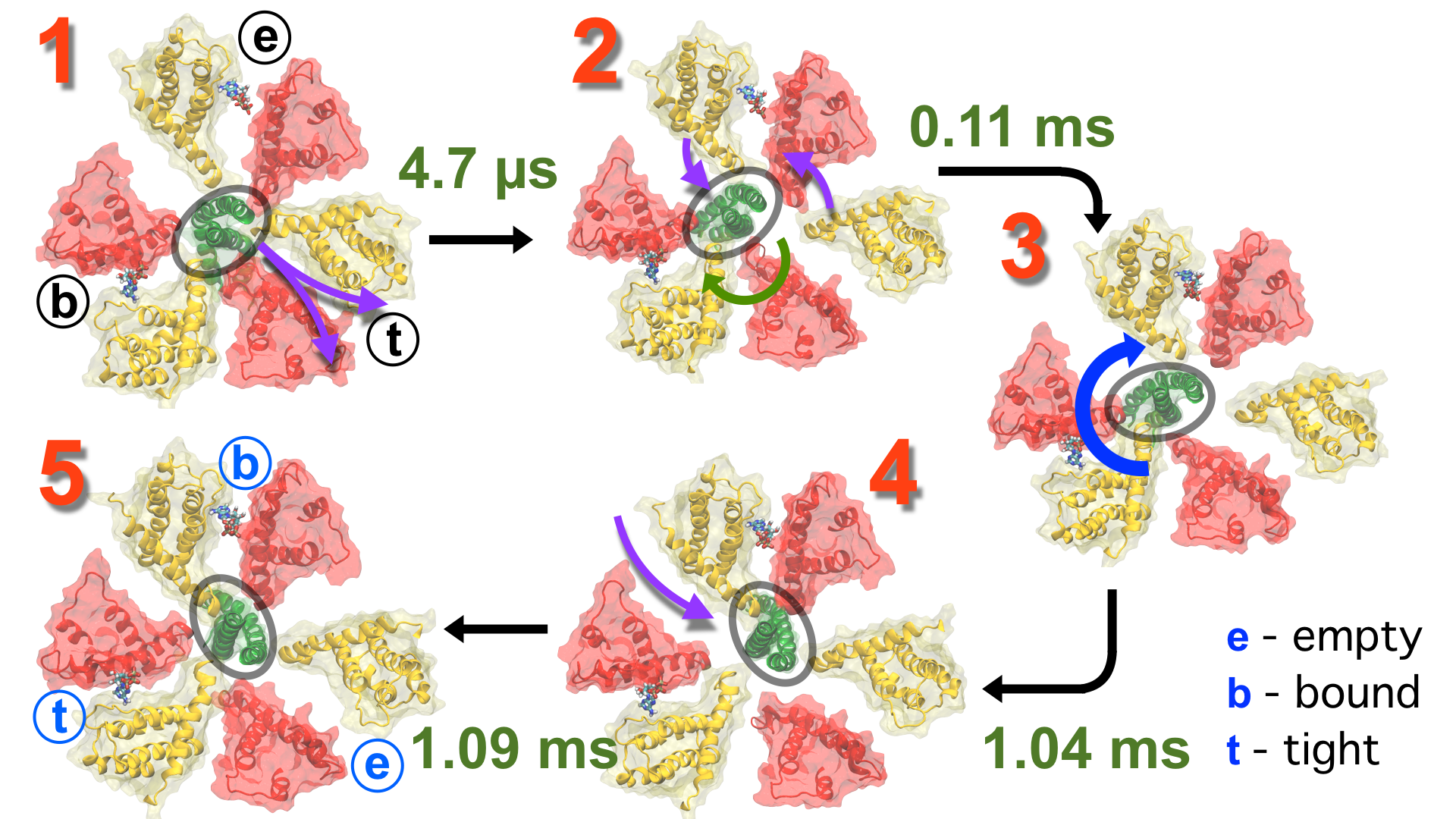
image size:
1.7MB
made with VMD
Living cells employ an intricate network of biochemical processes for the
conversion of solar energy or nutrients
into energy-rich Adenosine Tri Phosphate (ATP) molecules. From bacteria to
fungi, plants, and animals,
ATP serves as the universal energy currency of life, fueling the processes
cells need to survive and function.
Over the course of a day, an individual will typically use the equivalent of
his or her bodyweight in ATP; however, the human body carries only a small
amount of the molecule at any one time. That means cells must constantly
recycle or replenish the ATP molecules, relying on a highly efficient motor
protein called ATP synthase to do the job. Given its ubiquitous role in
photosynthesis and respiration, efficiency of the ATP synthase motor has been
a focus of intense biochemical investigations over the past three decades,
which included Boyer and Walker's Nobel Prize-winning contributions in 1997.
Yet, connection between the Chemistry of ATP dissociation and Physics of ATP
synthase motor-action remained elusive.
A recent study
based on molecular dynamics simulations with NAMD
reveals the working
principles of V-type ATP synthase in atomic resolution. Swiveling motions in
the protein ring were captured together with rubber band-like elasticity of
the motor's central stalk. This swiveling motion of the ring when paired with
the stalk absorbs about 75 percent of the energy released during ATP
hydrolysis, showcasing a molecular design that underlies the molecular motor's
remarkable energy-conversion efficiency.
More on ATPase here and
here.
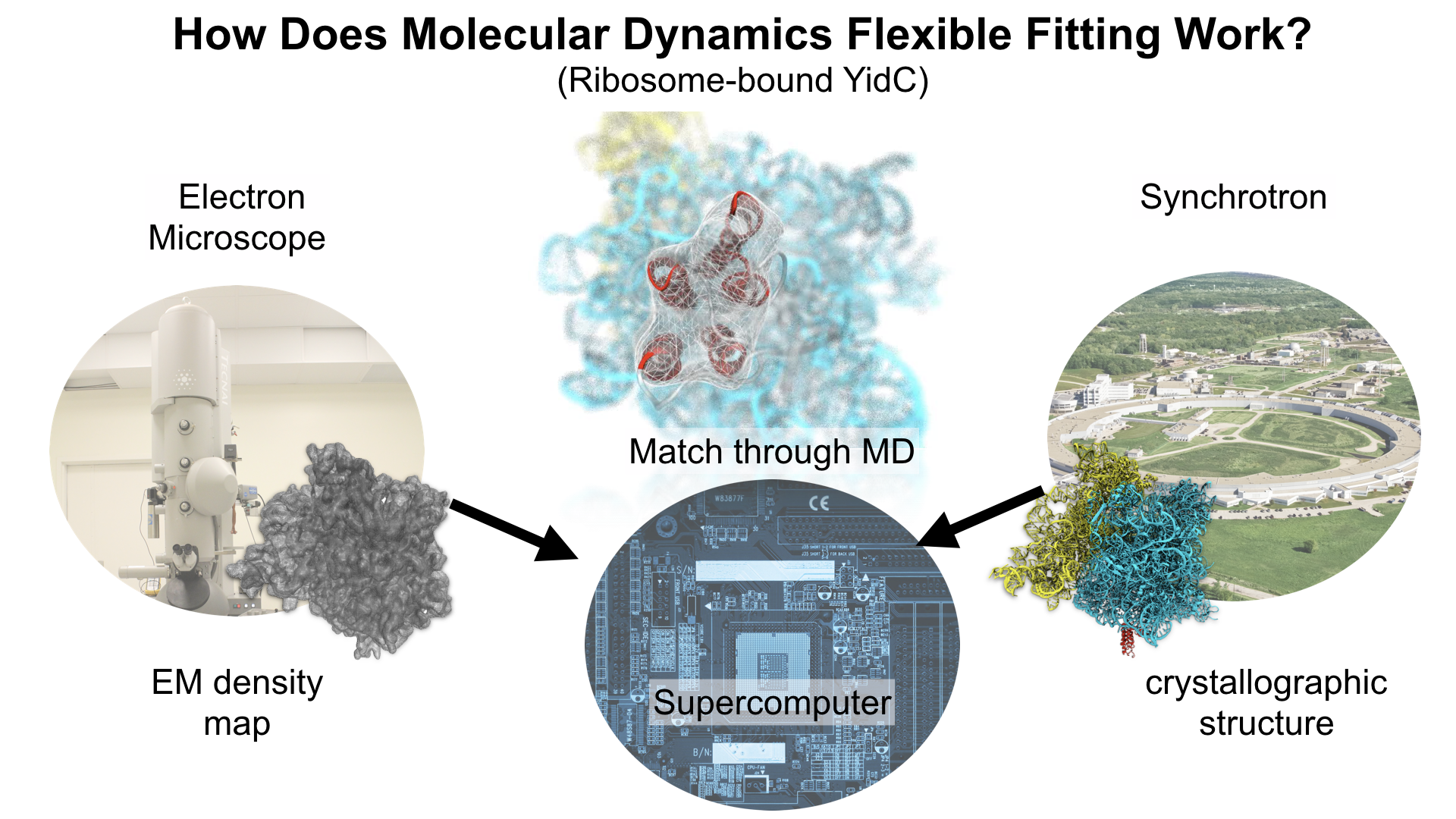
image size:
6.8MB
made with VMD
This year, the Royal Swedish Academy of Sciences awarded the Nobel Prize in Chemistry to
Jacques Dubochet,
Joachim Frank, and
Richard Henderson
"for developing cryo-electron microscopy for the high-resolution structure determination of biomolecules in solution".
We are pleased to celebrate this great triumph for structural biology along with the well-deserved recognition of the Center's long-time collaborator and friend, Joachim Frank.
Our center has a long tradition in developing computational methods that enable scientists to build atomistic models of biomolecules.
Molecular Dynamics Flexible Fitting (MDFF),
a method developed in close collaboration with Joachim Frank and his group,
reconciles high resolution data from X-ray crystallography and functional information from cryo-electron microscopy (cryo-EM).
MDFF utilizes molecular dynamics to "naturally" fit each atom into a cryo-EM map.
In less than a decade since its development, MDFF has proved instrumental in studying biomolecular systems.
A selected list of publications employing MDFF both by our group and others can be found
here.
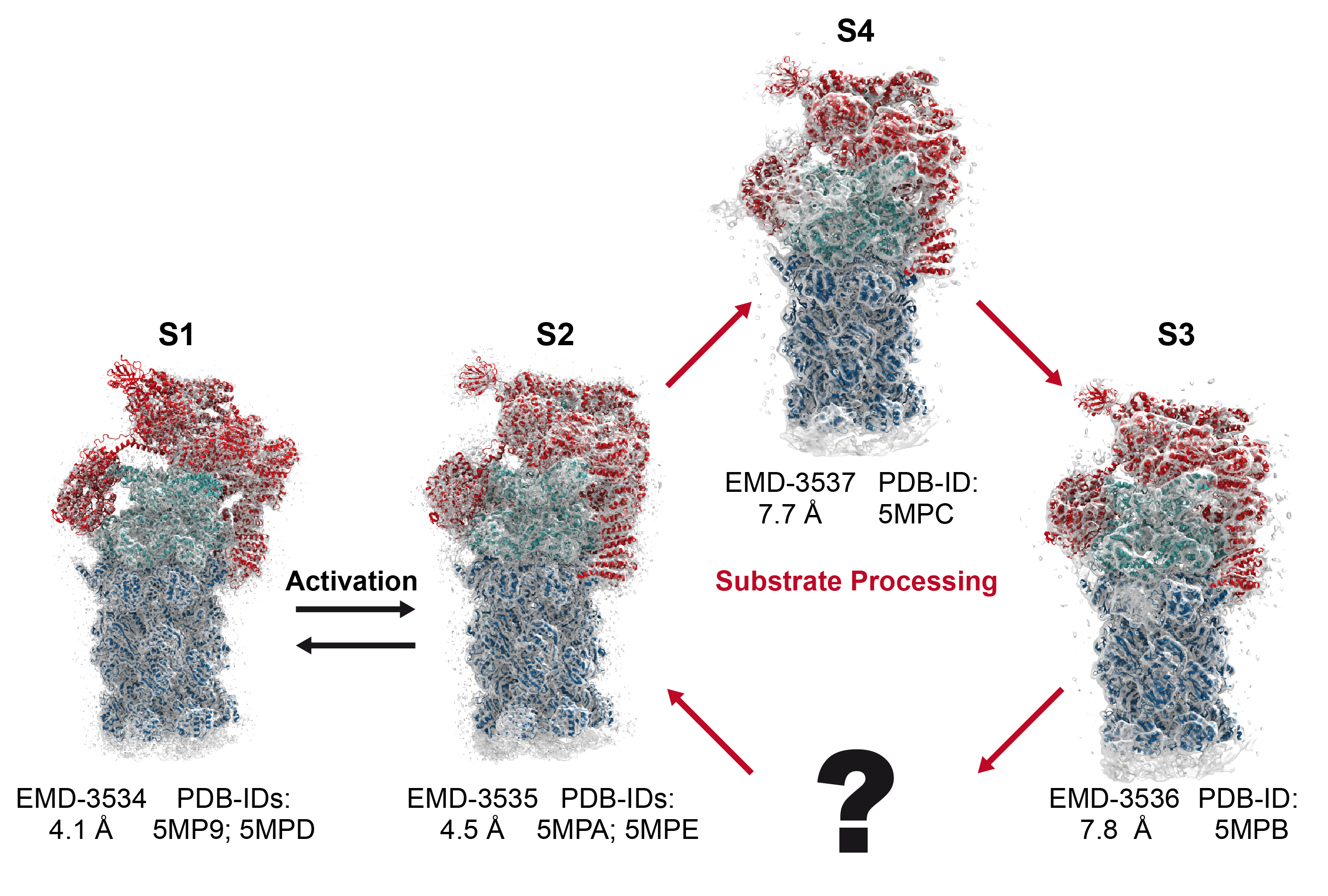
Protein recycling is a key process crucial to a wide spectrum of regulatory processes
within living cells. The executive player in this process is a molecular machine called
the proteasome, which both unfolds and chops superfluous proteins into smaller pieces
that will be used as raw building materials for new proteins. Given its critical role,
the proteasome is involved in multiple human diseases, and it serves as a perfect target
for a plethora of different drugs, most prominently, those commonly used in chemotherapy
of cancer. With the goal of understanding how these drugs work and paving the way for
designing even better drugs with less side effects, a
recent
collaborative study with
W. Baumeister (MPI Munich)
combined molecular dynamics flexible fitting (MDFF)
with de novo structure prediction algorithms to derive
four structural models from cryo-electron microscopy densities
of the proteasome in different conformational states. These models provide the first
atomic insights as to how ATP hydrolysis in the engine of the proteasome (cyan) unwinds
proteins and steers them towards the degradation chamber (red). More information is available on our
proteasome website. Easy access to our modeling techniques is
provided through QwikMD.
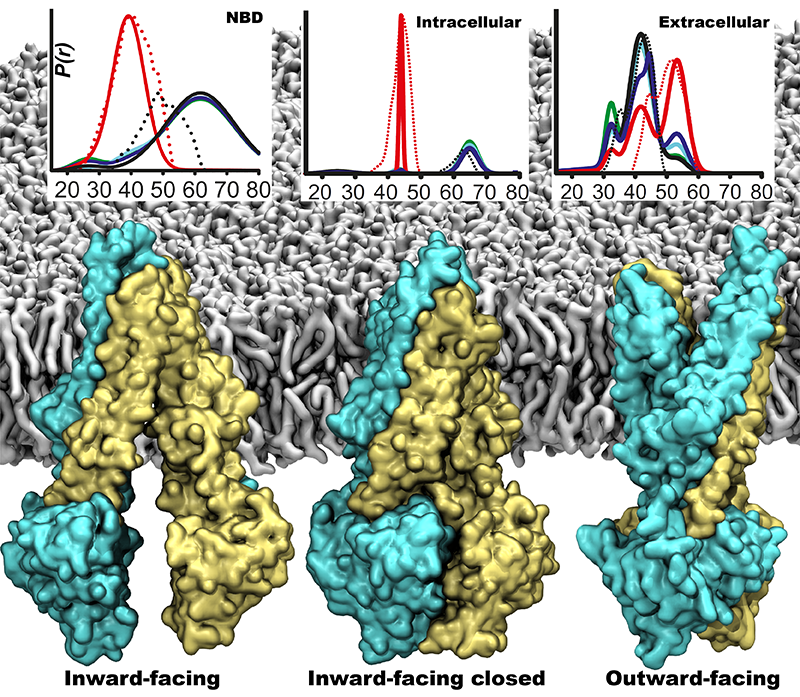
image size:
358.3KB
made with VMD
One of the most common mechanisms by which cancer and microbial cells develop resistance
against chemotherapeutic agents is to express a large number of specialized transporter proteins
in their cellular membrane that use the universal cellular energy of ATP to actively pump the drug
molecules to the outside. P-glycoprotein, a prominent member of such molecular "vacuum cleaners" and
responsible for multidrug resistance (MDR) in a wide variety of cancer types, accomplishes its role by
undergoing large-scale structural transitions in the cellular membrane through
which it effectively moves drug molecules from one side of the membrane to the other.
In a recent collaborative publication in Nature
with leading experimental groups
at Vanderbilt and Virginia, and employing advanced molecular modeling and
simulation techniques implemented in NAMD, a robust structural model was developed for
the unknown outward-facing state of P-glycoprotein,
allowing a full structural description of the transport cycle, and a novel mode of energy transduction.
Further details of the study can be found here.
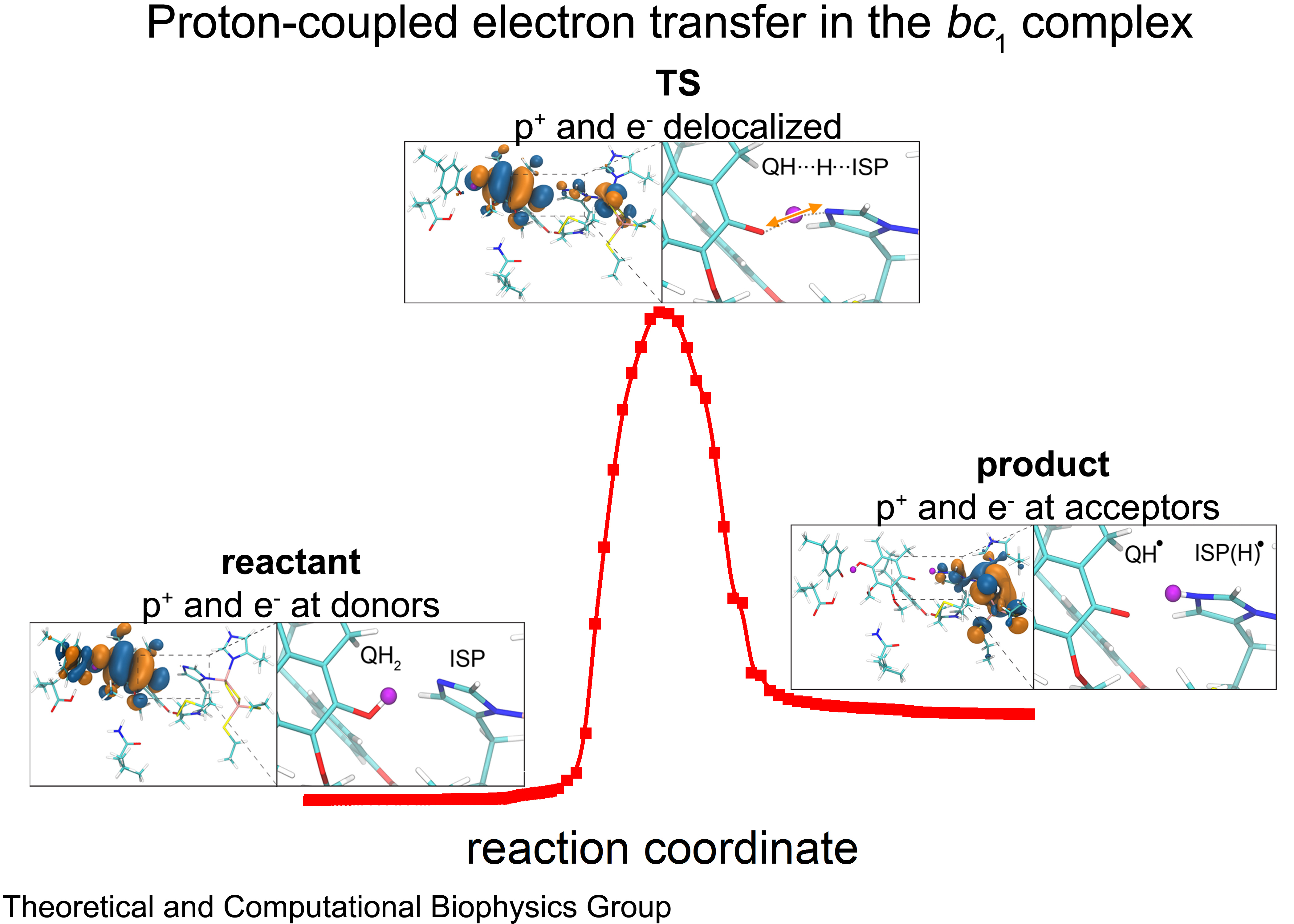
image size:
1.5MB
made with VMD
Cellular respiration and photosynthesis are the primary energy production mechanisms for sustaining life on earth.
Both processes use input energy (food or sunlight)
to drive coupled electron and proton transfer reactions,
thus replenishing the electrical charge of the cellular membrane,
which in turn is used to produce ATP - the universal fuel for all cellular activities.
A key step involved in both bacterial photosynthesis and mitochondrial respiration is
mediated by a specialized protein complex,
the bc1 complex,
which seizes the energy released from interconversion of quinol and quinone to pump protons across the bioenergetic membrane.
A recent study,
combining molecular simulations performed with NAMD and quantum chemical calculations,
unveiled the coupled nature of the proton and electron transfer reactions in the quinol binding site of the bc1 complex from a photosynthetic bacterium at an unprecedented level and described the role of key mechanistic elements.
More about the bc1 complex can be found here.
Journal of Physical Chemistry published a Memorial Issue ..." />

image size: 212.8KB
On April 20th, the Journal of Physical Chemistry published a Memorial Issue in honor of Klaus Schulten gathering more than 60 research articles. Klaus was an undisputed leader in theoretical and computational biophysics, recognized by his peers for his immense contribution to the field, and having devoted his entire career to establish the mechanisms that underlie cellular processes using the laws of physics. Originally planned to be a Festschrift to celebrate Klaus's achievements on his 70th birthday, the Memorial Issue initiative immediately triggered a unanimous positive response from friends, academic colleagues and longtime collaborators across the world. The many contributions assembled in the Memorial Issue of the Journal of Physical Chemistry lie at the confluence of theory and experiment, and cover a broad gamut of topics that were dear to Klaus, ranging from photosynthesis to molecular machines and membrane proteins. We gratefully acknowledge the many authors of the Memorial Issue, who enthusiastically accepted to pay one last homage to Klaus through contributions of very high scientific quality.
NAMD
provides major enhancements in ..." />
image size:
507.7KB
made with VMD
The 2.12 release of
the molecular dynamics program NAMD
provides major enhancements in performance, flexibility, and accuracy,
complementing the greatly enhanced usability provided by the
QwikMD GUI released in
VMD 1.9.3.
NVIDIA GPU-accelerated simulations with NAMD 2.12 are up to three times as
fast as 2.11, particularly for implicit solvent simulations and
single-node simulations of smaller systems.
NAMD 2.12 is also optimized for the new Intel Xeon Phi KNL processors found in
Argonne Theta,
NERSC Cori,
and
TACC Stampede 2.
NAMD 2.12 builds on the asynchronous multi-copy scripting capabilities introduced in
NAMD 2.11
with the ability to modify and reload the molecular structure,
enabling development of grand canonical and constant pH ensemble methods,
as well as an optional Python interface for advanced on-the-fly analysis.
Finally, NAMD 2.12 provides a complete, no-recompilation-needed
interface for hybrid QM/MM
with both the semi-empirical code MOPAC and the ab initio/DFT code ORCA.
More on new features in the 2.12 release of NAMD can be found
here.
NAMD is available free-of-charge as source code, precompiled binaries,
pre-installed at supercomputer centers, and now jointly with VMD as
one-click interactive molecular modeling
on the Amazon cloud.
VMD brings many
advances that help researchers prepare, ..." />


image size:
2.2MB
made with VMD
The latest release of VMD brings many
advances that help researchers prepare, analyze, and visualize
molecular simulations.
The new
QwikMD plugin
streamlines key simulation preparation and analysis tasks, and guides users
in the creation of reusable simulation workflows and protocols.
VMD now includes several advanced features for parallel analysis
and visualization of cellular-scale simulations, as
reported here,
and here.
VMD 1.9.3 strengthens collaboration between experimental and computational
biologists by supporting a broader range of experimental density map
image formats, such as those used in cryo-electron tomography.
Many updated plugins are included in VMD 1.9.3, including tools for
analysis of free energy perturbation simulations,
MDFF hybrid structure fitting,
ffTK force field parameterization,
and
normal mode analysis.
VMD 1.9.3 adds support for new hardware and operating system
platforms including
IBM OpenPOWER (ORNL Summit),
a variety of GPU-accelerated ARM SoCs,
the Amazon AWS EC2 cloud,
and most recently, the Intel Xeon Phi Knight's Landing many-core CPU (TACC Stampede 2, Argonne Theta).
The VMD 1.9.3 release adds stunning graphics produced using
interactive ray tracing using the latest multi-core CPUs and GPU accelerators,
enabling 360-degree panoramic movie rendering for VR headsets,
as
reported here,
and here.
Interactive ray tracing makes the task of getting a molecular
image "just right" much easier than ever before; it also enables
rendering of spectacular movies for communication of scientific results.
A VR movie rendering tutorial
assists users with the steps required in rendering and encoding
VR movies for upload to YouTube for display using VR headsets such
as Google Cardboard, Oculus Rift, and GearVR.
More details about VMD 1.9.3 features can be found
here.

image size:
792.2KB
made with VMD
ATP, a ubiquitously prevalent biomolecule, which is best known for being the principal energy source for a living cell, also plays a crucial role in inter-cellular communication, thus acting as a signaling molecule. One of the major receptors in this signaling cascade are the P2X receptors which are trimeric, non-selective cation channel activated by ATP and responsible for key processes such as muscle contraction, inflammatory response, pain, and even taste signal transduction. As a result of their extensive prevalence and important implications in human physiology, P2X receptors serve as important pharmacological targets for cardiovascular, neuronal, and inflammatory diseases.
In a recent collaborative study with experimental structural biologists, molecular dynamics simulations of a membrane-embedded model of a P2X receptor performed with NAMD were used to reveal intricate details of the ion permeation mechanism and pathway. Surprisingly, it was observed that one half of the ion permeation pathway is composed of lipids on one side and of the protein residues on the other side, a novel design for an ion translocation pore. The study demonstrates yet another active functional role for lipids in membrane protein function, further emphasizing the importance of lipid protein interactions in biological processes.
More details can be found here.
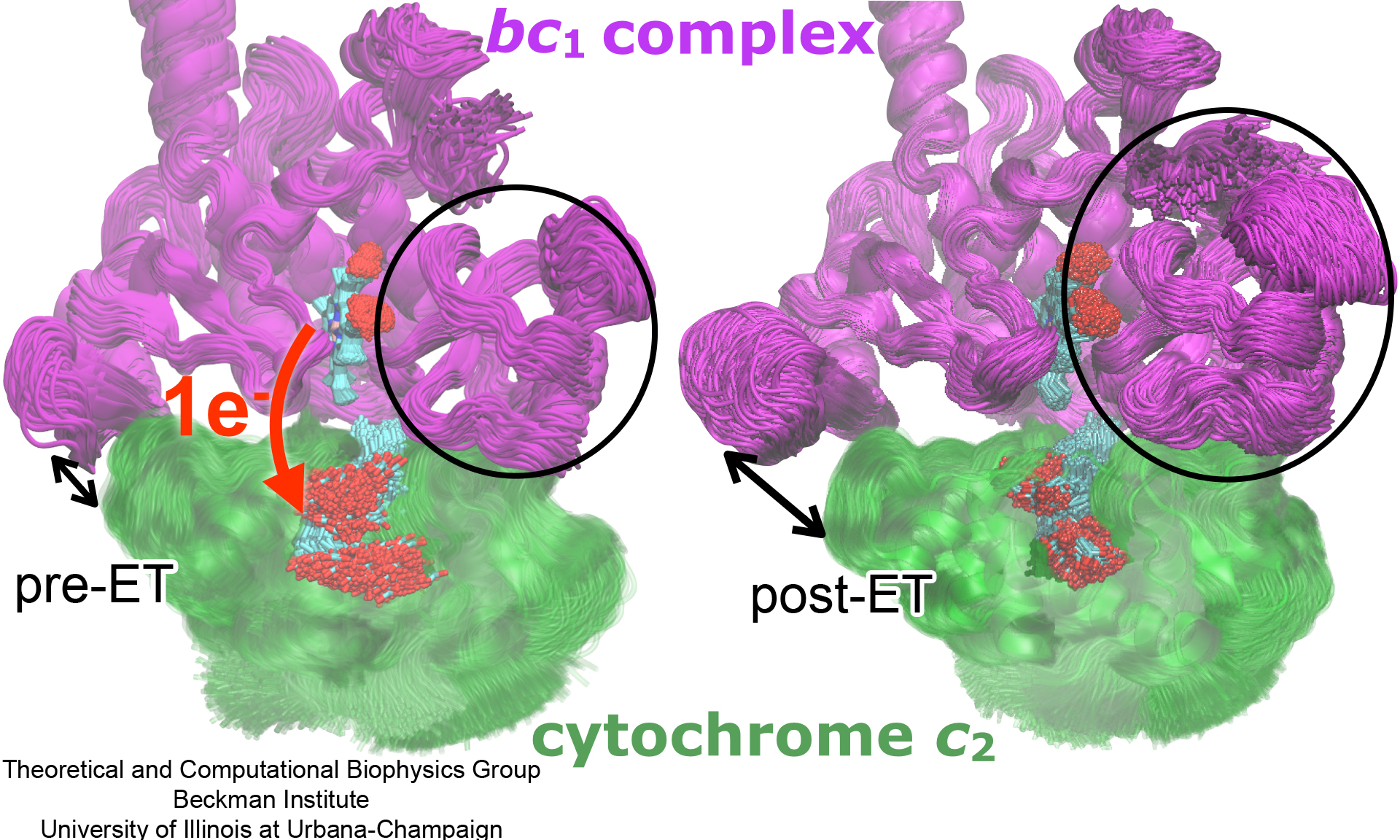
image size:
1.6MB
made with VMD
Photosynthetic organisms have been optimized by over two billion years of evolution into energy-harvesting machines that surpass the efficiency of man-made solar devices.
Employing a network of hundreds of proteins, these organisms transform energy from the incident sunlight into ATP molecules - the universal fuel for sustaining cellular activities.
A key step in the conversion of solar energy into ATP involves shuttling of negative charges or electrons between widely separated sites on the cell membrane.
This membrane-wide charge transportation is accomplished by the cytochrome c family of proteins.
Cytochrome c finds and docks to an electron donor protein, accepts the electron and unbinds as a result of it, and afterwards finds an electron acceptor protein, docks to it to deliver the electron and unbinds to repeat the sequence.
Given its ubiquitous role in photosynthesis and respiration, the recognition, binding and unbinding mechanism of cytochrome c have been a focus of intense biochemical investigations over the past three decades.
However, little is deciphered on the details of cytochrome c activity.
A recent study based on molecular dynamics simulations with NAMD reveals the working principles of cytochrome c in atomic resolution.
The calculations suggest that electrostatic forces drive the cytochrome c binding, and enable membrane-wide electron transfer.
More about the cytochrome c protein can be found here.


