Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
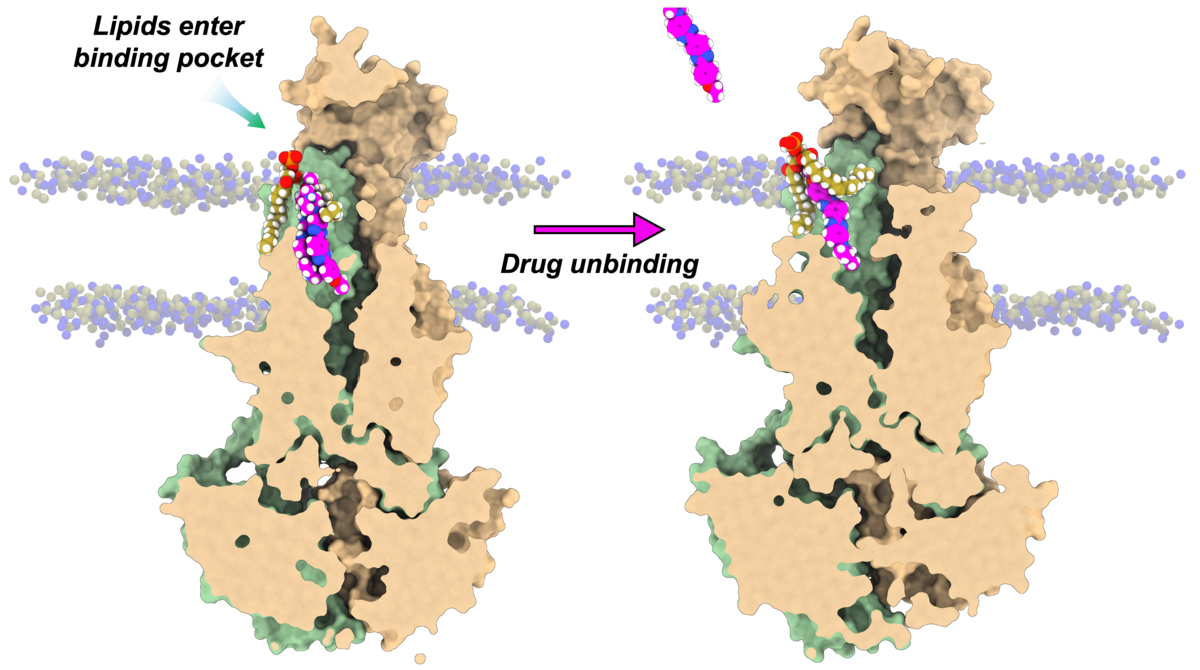
ABC exporters are critical molecular machines that use ATP to pump xenobiotic molecules such as drugs across cell membranes. They often drive multidrug resistance, most prominently in cancer. However, the molecular mechanism of their function remains elusive. Building on previous work revealing how lipids stabilize the transporter structure, and in collaboration with the Mchaourab lab at Vanderbilt, the Resource used VMD 2.0 to model partially resolved cryo-EM lipids in an ABC exporter structure and simulated them with NAMD 3.0. The results provide yet another piece of evidence for competition between bound drugs and lipids. For more details, see our recent publication in Nature Communications.

image size:
247.0KB
Workshop photo
The 64th Hands-on Workshop on Computational Biophysics took place at Auburn University between 6/22 - 6/26. This year we were able to host approximately 30 participants from all over the world! The workshop covered topics ranging from the basics of molecular dynamics to advanced free energy calculation and enhanced sampling methods, as well as molecular visualization, analysis, 3D rendering, and animations. The workshop employed the latest VMD 2.1 alpha pre-release! Stay tuned for upcoming workshops in the future!
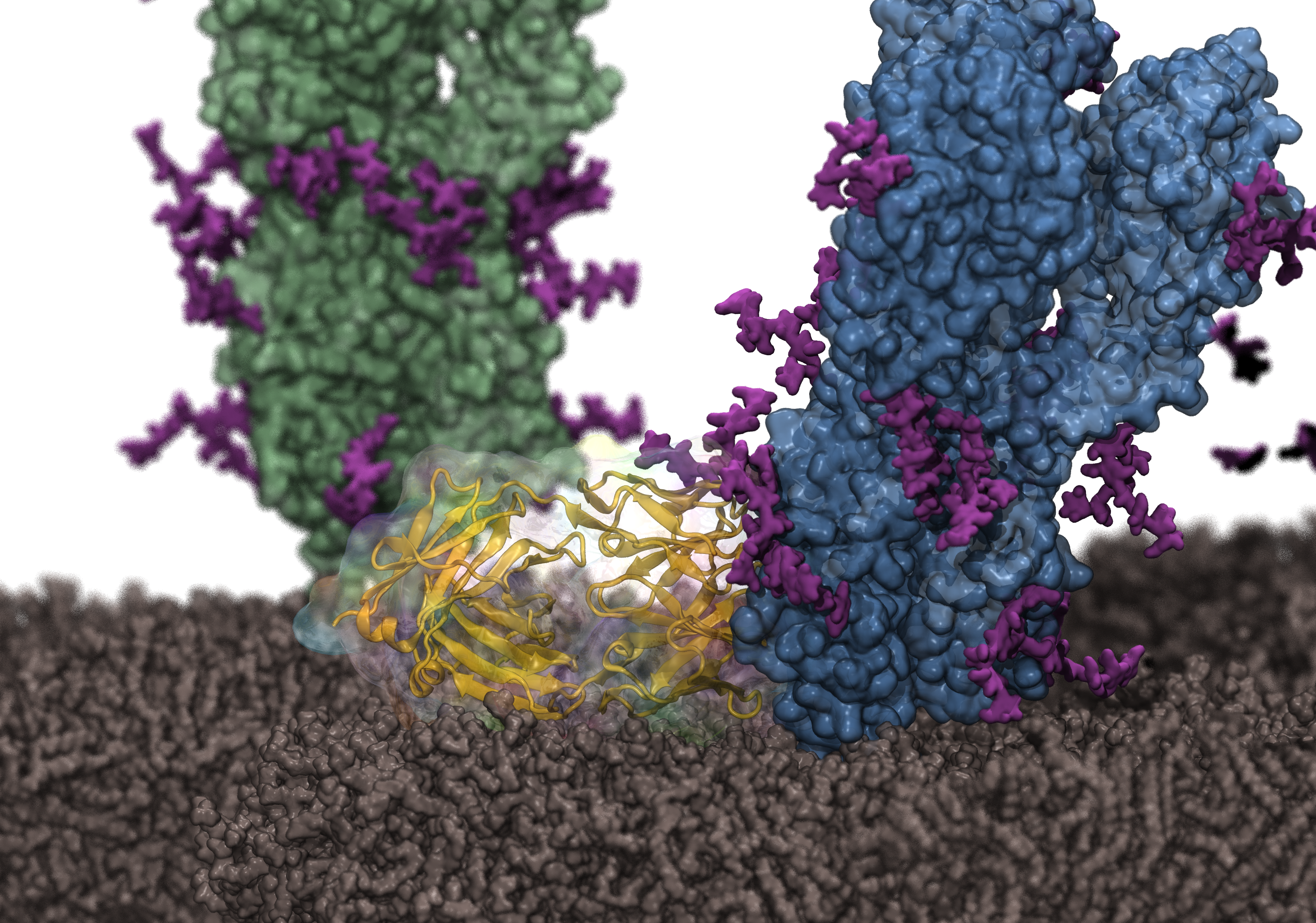
Influenza hemagglutinin (HA) is the most abundant surface antigen of the virus. Since there are 18 subtypes of HA with different antigenicity, there is a strong need to develop broadly neutralizing antibodies targeting multiple subtypes. In collaboration with the Wu Lab at the University of Illinois, Resource researchers reconstructed the structure of the complex between HA and a broadly neutralizing antibody on the viral membrane and simulated it with NAMD. The simulation results revealed membrane interactions for the antibody, which were then confirmed by mutagenesis experiments. The results are made recently available as a cover article in Structure and provide a foundation for rational design of more effective antibodies.
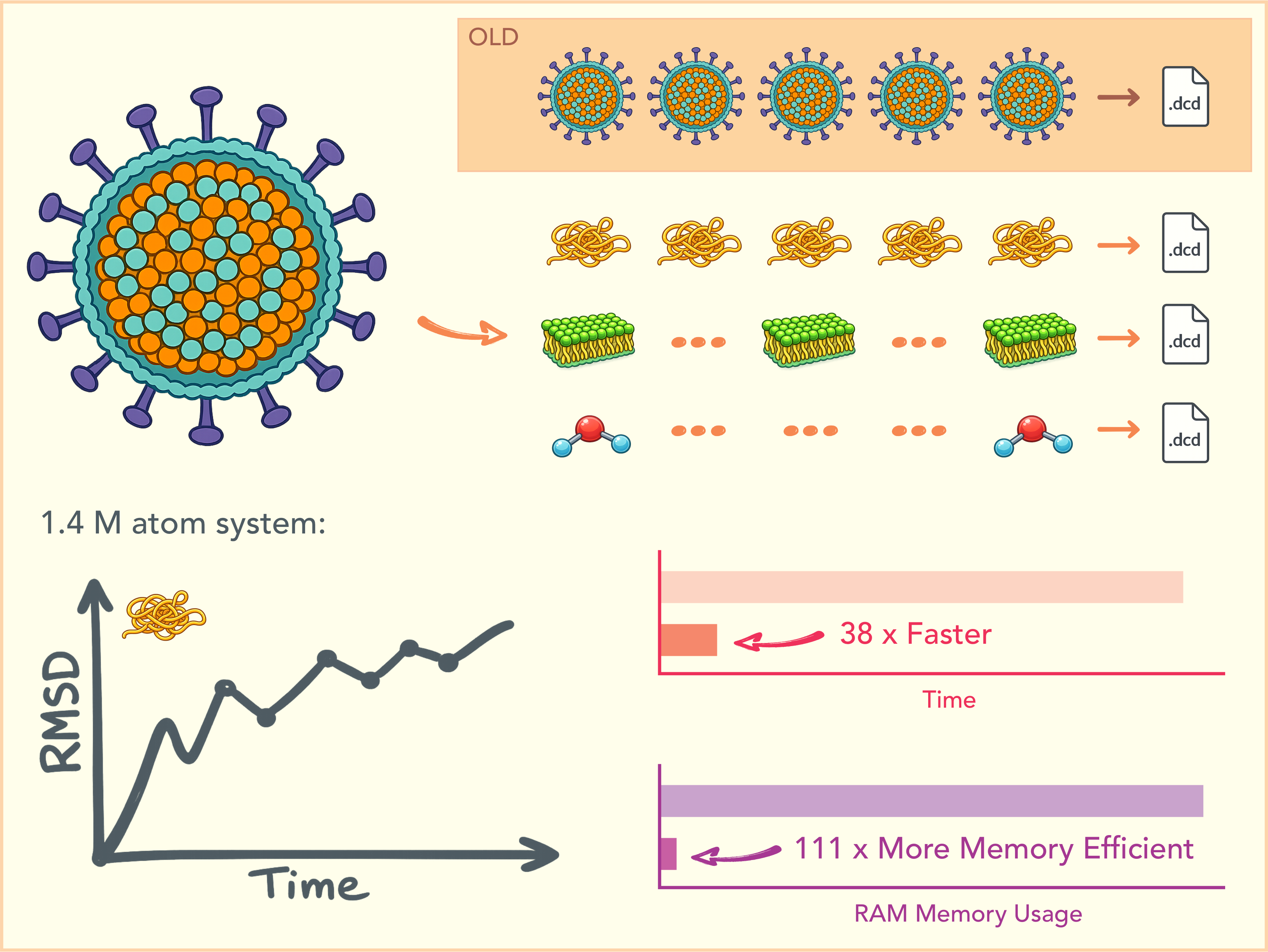
image size: 1.5MB
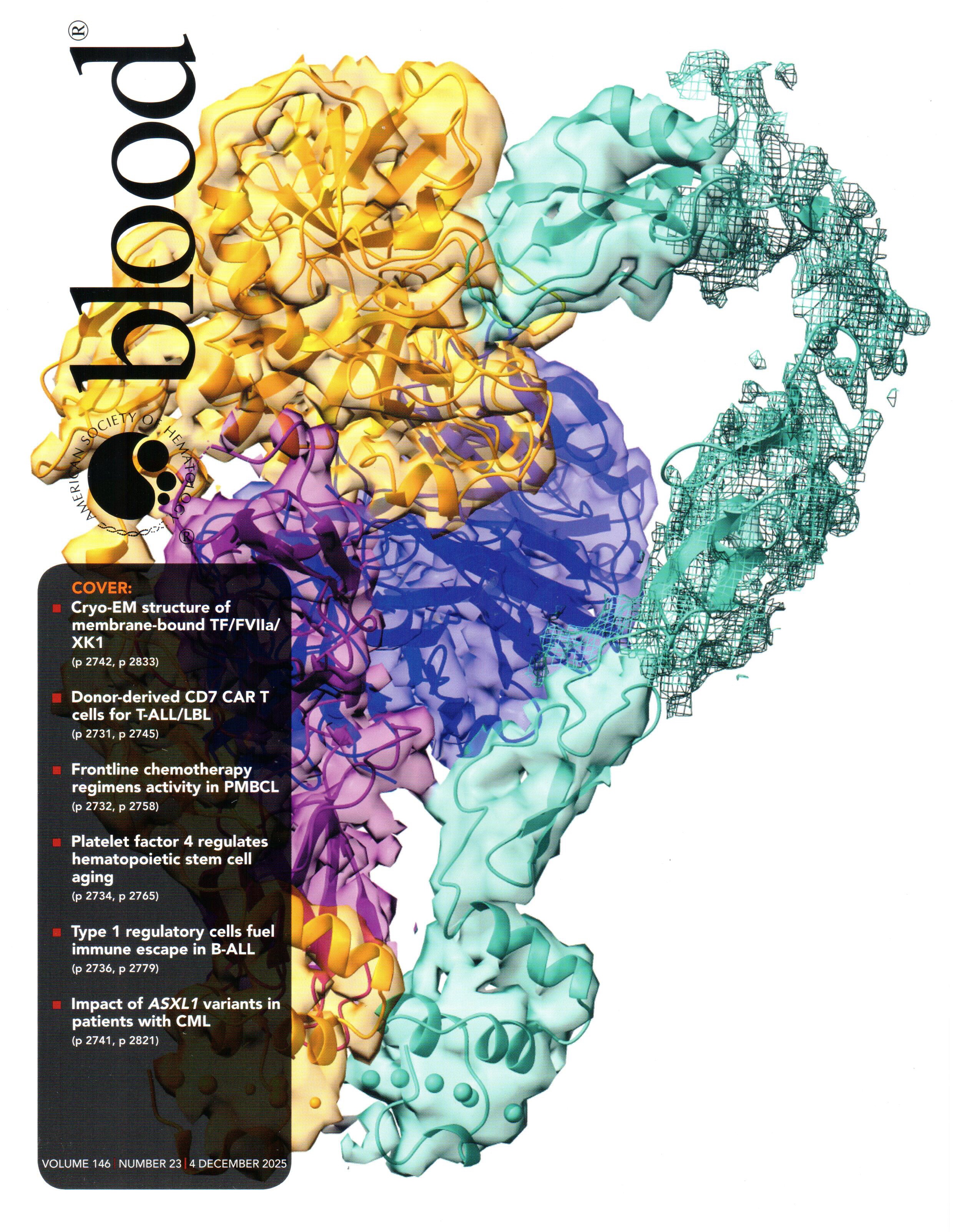
Activation of coagulation factor X (FX) is arguably the most important step in the formation of blood clots. When tissues are damaged by external injury, FX activation is initiated by the formation of a complex between FVIIa and tissue factor, which binds and activates FX after anchoring into negatively charged cellular membranes. This key step involves the formation of a tripartite complex on the membrane, which was earlier modeled by Resource researchers and published in Blood Advances. In collaboration with the Ohi lab and Morrissey lab at the University of Michigan, the previous model is now largely confirmed by the first cryo-EM structure of the complex on the membrane, a study reported as a cover story in Blood.


