GPU Acceleration of Molecular Modeling Applications


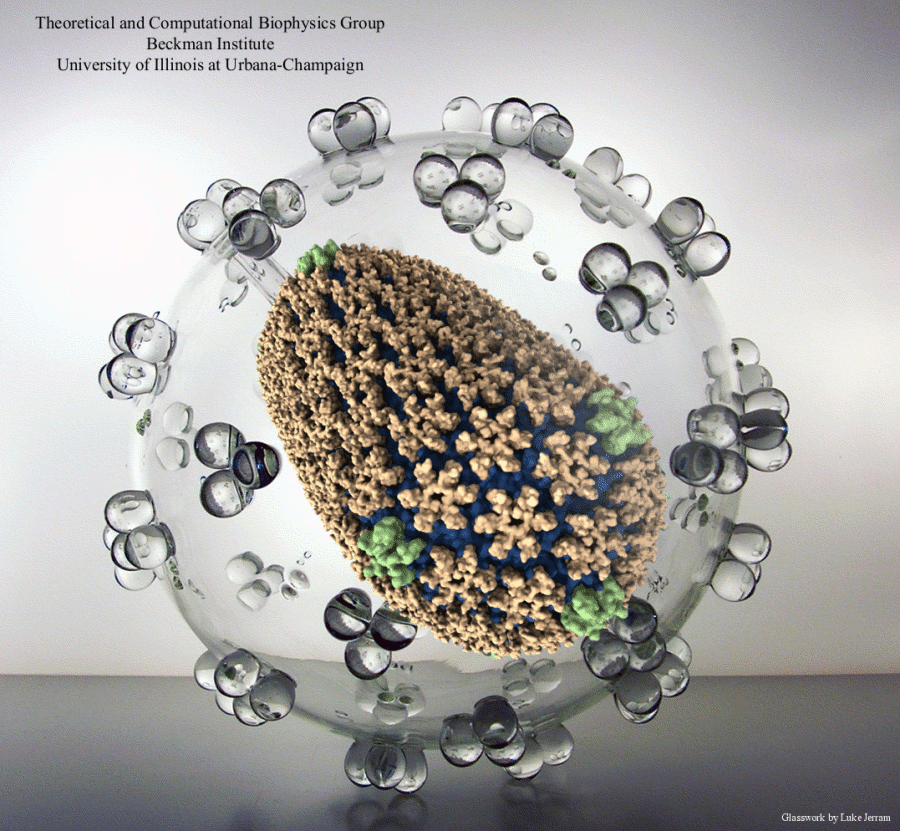
Elusive HIV-1 Capsid Structure Determination Accelerated by GPUs
Molecular Dynamics

Power Profiling

Multi-Resolution Molecular Surface Visualization
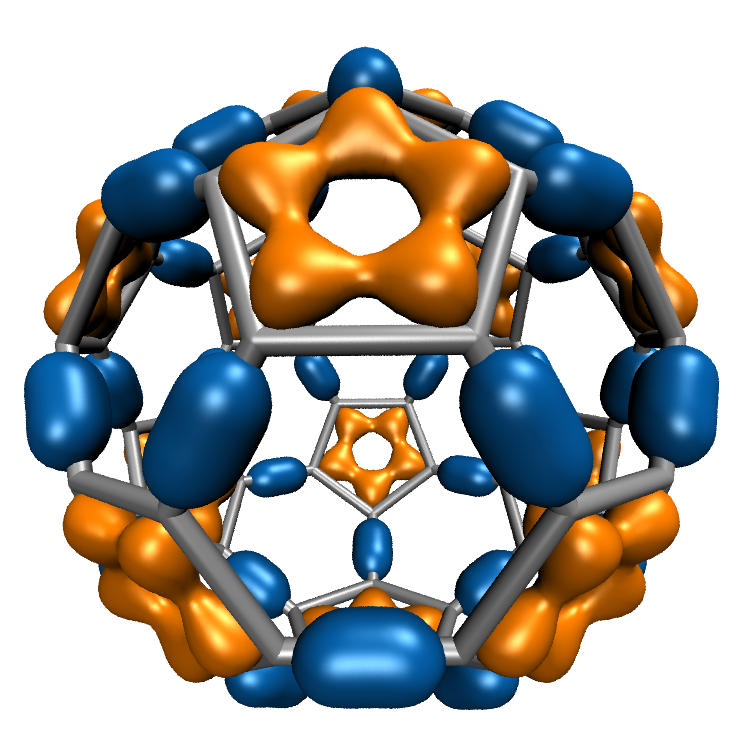
Molecular Orbital Display
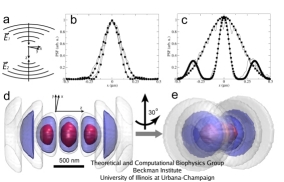
Visualization of molecular orbitals (MOs) is important for analyzing the results of quantum chemistry simulations. The functions describing the MOs are computed on a three-dimensional lattice, and the resulting data can then be used for plotting isocontours or isosurfaces for visualization as well as for other types of analyses. Existing software packages that render MOs perform calculations on the CPU and require runtimes of tens to hundreds of seconds depending on the complexity of the molecular system.
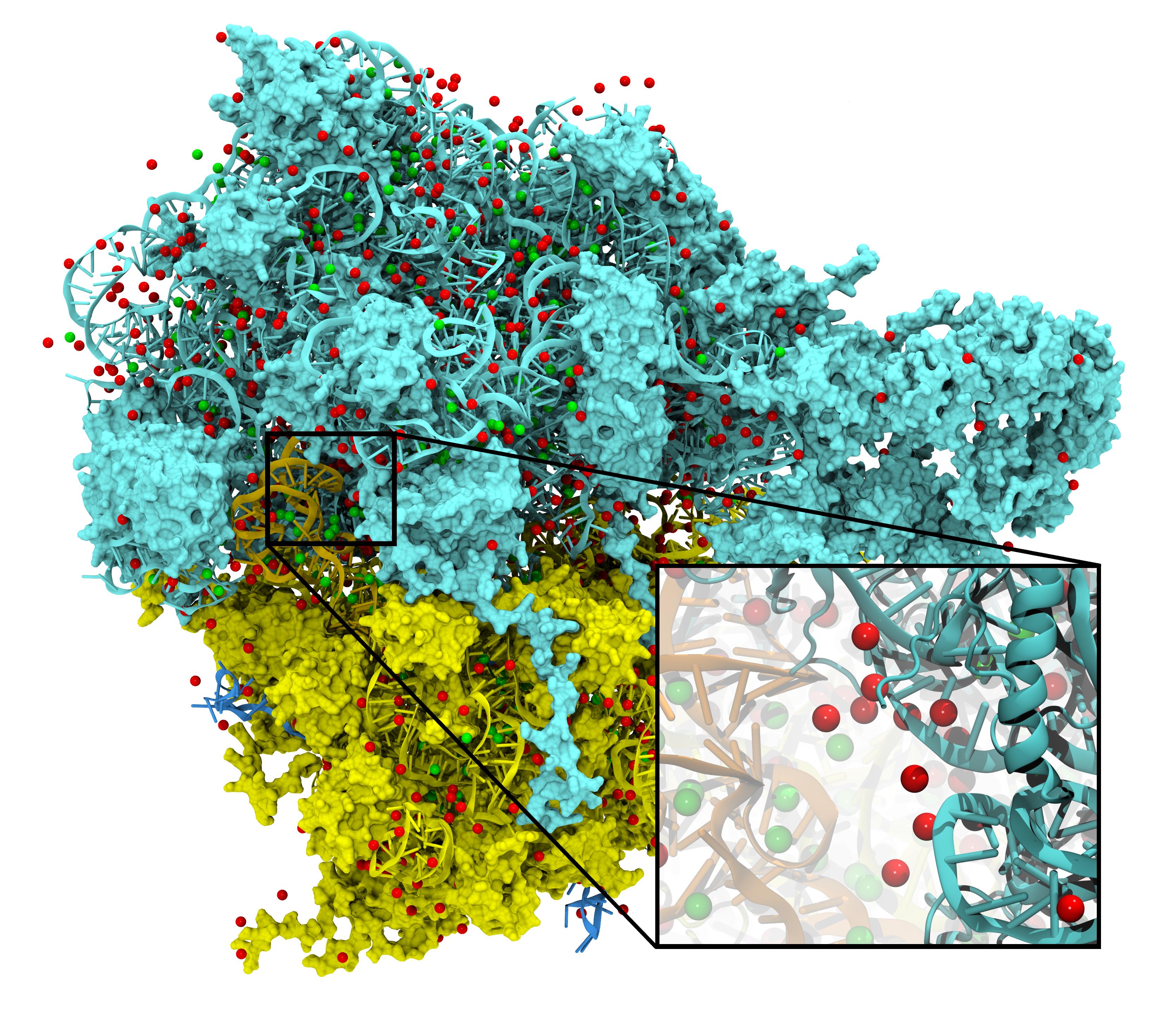
Ion Placement
To best reproduce physiological conditions, molecular dynamics simulations must be run in the presence of appropriate ions. Generally such simulations are performed in the presence of sodium chloride, although in some cases (such as simulations including nucleic acid) other ions such as magnesium are necessary. Although many tools such as the VMD Autoionize plugin can place a random distribution of ions, molecules requiring counterions for their stability are better treated using ion placement methods which take the electrostatics of the solute into account. One method for doing this is to place important counterions at minima in the electrostatic potential field generated by the biomolecule of interest, iteratively updating the potential field after each ion is placed.

While this method of ion placement is simple and computes ion positions matched to the specific target molecule, it can be very computationally demanding for large structures because it requires calculation of the electrostatic potential at all points on a high-resolution 3-D lattice in the neighborhood of the solute. Coulomb-based ionization of very large structures such as viruses could require several days even using moderately sized clusters of computers. However, the calculation of a function on a lattice where all points are independent is an ideal application for GPU acceleration, and as recently reported in the Journal of Computational Chemistry, the use of GPUs to accelerate Coulomb-based ion placement leads to speedups of 100 times or more, allowing large structures to be properly ionized in less than an hour on a single desktop computer.
|

|
|
|
|
The direct summation of the Coulomb potential from all atoms to every lattice point requires computational work that grows quadratically, proportional to the product of the number of atoms and the number of lattice points. An algorithmic enhancement known as multilevel summation uses hierarchical interpolation of softened pairwise potentials from lattices of increasing coarseness to compute an approximation to the Coulomb potential. The amount of computational work for multilevel summation grows linearly, proportional to the sum of the number of atoms and the number of lattice points. Our reported GPU-assisted implementation of this method further reduces the time of obtaining large ionized structures to just a few minutes on a single desktop computer. The accuracy of the implementation is sufficient (with an average difference from the direct approach demonstrated to be in the range of 0.025% to 0.037%) to permit identical ion placement as the direct summation approach for small test molecules and nearly identical results for the ribosome.
The GPU-accelerated Coulomb potential calculation can be directly applied to calculate time-averaged electrostatic potentials from molecular dynamics simulations. As we reported, a VMD calculation of the electrostatic potential for one frame of a molecular dynamics simulation of the ribosome takes 529 seconds on a single GPU, as opposed to 5.24 hours on a single CPU core. A multilevel summation calculation for a single frame requires 67 seconds on one GPU.
Multi-GPU Coulomb Summation

The direct Coulomb summation algorithm implemented in VMD is an exemplary case for multi-GPU acceleration. The scaling efficiency for direct summation across multiple GPUs is nearly perfect -- the use of 4 GPUs delivers almost exactly 4X performance increase. A single GPU evaluates up to 39 billion atom potentials per second, performing 290 GFLOPS of floating point arithmetic. With the use of four GPUs, total performance increases to 157 billion atom potentials per second and 1.156 TFLOPS of floating point arithmetic, for a multi-GPU speedup of 3.99 and a scaling efficiency of 99.7%, as recently reported. To match this level of performance using CPUs, hundreds of state-of-the-art CPU cores would be required, along with their attendant cabling, power, and cooling requirements. While only one of the first steps in our exploration of the use of multiple GPUs, this result clearly demonstrates that it is possible to harness multiple GPUs in a single system with high efficiency.
Fluorescence Microphotolysis
Software
- NAMD Molecular Dynamics
- VMD Molecular Visualization
- GPU Programming for Molecular Modeling Worksop, 2010.
- GPU Computing Gems Vol. 1 Source Code:
- GPUComputing.Net
- GPGPU.org
- Khronos Group, OpenCL specifications and software
- Download the AMD/ATI OpenCL toolkit and drivers
- Download the NVIDIA CUDA and OpenCL toolkits and drivers
Book Chapters
 |
|
|
|
|
 |
|
 |
|
 |
|
 |
|
|
|
 |
|
Publications
- Scalable Molecular Dynamics with NAMD on the Summit System. Bilge Acun, David J. Hardy, Laxmikant V. Kale, Ke Li, James C. Phillips, and John E. Stone. IBM Journal of Research and Development, 2018.
- Best Practices in Running Collaborative GPU Hackathons: Advancing Scientific Applications with a Sustained Impact. Sunita Chandrasekaran, Guido Juckeland, Meifeng Ling, Matthew Otten, Dirk Pleiter, John E. Stone, Juan Lucio-Vega, Michael Zingale, and Fernanda Foertter. Computing in Science and Engineering 20(4):95-106, 2018.
Publications Database - NAMD goes quantum: An integrative suite for hybrid simulations. Marcelo C. R. Melo, Rafael C. Bernardi, Till Rudack, Maximilian Scheurer, Christoph Riplinger, James C. Phillips, Julio D. C. Maia, Gerd B. Rocha, João V. Ribeiro, John E. Stone, Frank Nesse, Klaus Schulten, and Zaida Luthey-Schulten. Nature Methods, 15:351-354, 2018.
- The OLCF GPU Hackathon Series: The Story Behind Advancing Scientific Applications with a Sustained Impact. Sunita Chandrasekaran, Guido Juckeland, Meifeng Ling, Matthew Otten, Dirk Pleiter, John E. Stone, Juan Lucio-Vega, Michael Zingale, and Fernanda Foertter. The 2017 Workshop on Education for High Performance Computing (EduHPC'17), 2017.
- Challenges of Integrating Stochastic Dynamics and Cryo-electron Tomograms in Whole-cell Simulations. Tyler M. Earnest, Reika Watanabe, John E. Stone, Julia Mahamid, Wolfgang Baumeister, Elizabeth Villa, and Zaida Luthey-Schulten.
J. Physical Chemistry B, 121(15): 3871-3881, 2017.
Publications Database - Early experiences porting the NAMD and VMD molecular simulation and analysis software to GPU-accelerated OpenPOWER platforms. John E. Stone, Antti-Pekka Hynninen, James C. Phillips, and Klaus Schulten. International Workshop on OpenPOWER for HPC (IWOPH'16), pp. 188-206, 2016.
- Atomic detail visualization of photosynthetic membranes with GPU-accelerated ray tracing. John E. Stone, Melih Sener, Kirby L. Vandivort, Angela Barragan, Abhishek Singharoy, Ivan Teo, Joao V. Ribeiro, Barry Isralewitz, Bo Liu, Boon Chong Goh, James C. Phillips, Craig MacGregor-Chatwin, Matthew P. Johnson, Lena F. Kourkoutis, C. Neil Hunter, and Klaus Schulten. Parallel Computing, 55:17-27, 2016.
- Immersive molecular visualization with omnidirectional stereoscopic ray tracing and remote rendering. John E. Stone, William R. Sherman, and Klaus Schulten. 2016 IEEE International Parallel and Distributed Processing Symposium Workshop (IPDPSW), pp. 1048-1057, 2016.
- High performance molecular visualization: In-situ and parallel rendering with EGL. John E. Stone, Peter Messmer, Robert Sisneros, and Klaus Schulten. 2016 IEEE International Parallel and Distributed Processing Symposium Workshop (IPDPSW), pp. 1014-1023, 2016.
- Evaluation of emerging energy-efficient heterogeneous computing platforms for biomolecular and cellular simulation workloads. John E. Stone, Michael J. Hallock, James C. Phillips, Joseph R. Peterson, Zaida Luthey-Schulten, and Klaus Schulten. 2016 IEEE International Parallel and Distributed Processing Symposium Workshop (IPDPSW), pp. 89-100, 2016.
- Chemical visualization of human pathogens: the Retroviral Capsids. Juan R. Perilla, Boon Chong Goh, John Stone, and Klaus Schulten. Proceedings of the 2015 ACM/IEEE Conference on Supercomputing, 2015. (4 pages).
- Multilevel summation method for electrostatic force evaluation. David J. Hardy, Zhe Wu, James C. Phillips, John E. Stone, Robert D. Skeel, and Klaus Schulten. Journal of Chemical Theory and Computation, 11:766-779, 2015.
-
Runtime and Architecture Support for Efficient Data Exchange in Multi-Accelerator Applications,
Javier Cabezas, Isaac Gelado, John E. Stone,
Nacho Navarro, David B. Kirk, and Wen-mei Hwu.
IEEE Transactions on Parallel and Distributed Systems, 26(5):1405-1418, 2015.
Publications Database - Visualization of energy conversion processes in a light harvesting organelle at atomic detail. Melih Sener, John E. Stone, Angela Barragan, Abhishek Singharoy, Ivan Teo, Kirby L. Vandivort, Barry Isralewitz, Bo Liu, Boon Chong Goh, James C. Phillips, Lena F. Kourkoutis, C. Neil Hunter, and Klaus Schulten. In Proceedings of the International Conference on High Performance Computing, Networking, Storage and Analysis, SC '14. IEEE Press, 2014. (4 pages).
- GPU-accelerated analysis and visualization of large structures solved by molecular dynamics flexible fitting. John E. Stone, Ryan McGreevy, Barry Isralewitz, and Klaus Schulten. Faraday Discussions, 169:265-283, 2014.
- Simulation of reaction diffusion processes over biologically relevant size and time scales using multi-GPU workstations. Michael J. Hallock, John E. Stone, Elijah Roberts, Corey Fry, and Zaida Luthey-Schulten. Parallel Computing, 40:86-99, 2014.
- Unlocking the full potential of the Cray XK7 accelerator. Mark D. Klein and John E. Stone. In Cray User Group Conference. Cray, May 2014.
- GPU-accelerated molecular visualization on petascale supercomputing platforms. John E. Stone, Kirby L. Vandivort, and Klaus Schulten. In Proceedings of the 8th International Workshop on Ultrascale Visualization, UltraVis '13, pp. 6:1-6:8, New York, NY, USA, 2013. ACM.
- Early experiences scaling VMD molecular visualization and analysis jobs on Blue Waters. John E. Stone, Barry Isralewitz, and Klaus Schulten. In Extreme Scaling Workshop (XSW), 2013, pp. 43-50, Aug. 2013.
- Lattice microbes: High-performance stochastic simulation method for the reaction-diffusion master equation. Elijah Roberts, John E. Stone, and Zaida Luthey-Schulten. Journal of Computational Chemistry, 34:245-255, 2013.
- Fast visualization of Gaussian density surfaces for molecular dynamics and particle system trajectories. Michael Krone, John E. Stone, Thomas Ertl, and Klaus Schulten. In EuroVis - Short Papers 2012, pp. 67-71, 2012.
- Fast analysis of molecular dynamics trajectories with graphics processing units-radial distribution function histogramming. Benjamin G. Levine, John E. Stone, and Axel Kohlmeyer. Journal of Computational Physics, 230:3556-3569, 2011.
- Immersive molecular visualization and interactive modeling with commodity hardware. John E. Stone, Axel Kohlmeyer, Kirby L. Vandivort, and Klaus Schulten. Lecture Notes in Computer Science, 6454:382-393, 2010.
- Quantifying the impact of GPUs on performance and energy efficiency in HPC clusters. Jeremy Enos, Craig Steffen, Joshi Fullop, Michael Showerman, Guochun Shi, Kenneth Esler, Volodymyr Kindratenko, John E. Stone, and James C. Phillips. In International Conference on Green Computing, pp. 317-324, 2010.
- GPU-accelerated molecular modeling coming of age. John E. Stone, David J. Hardy, Ivan S. Ufimtsev, and Klaus Schulten. Journal of Molecular Graphics and Modelling, 29:116-125, 2010.
- OpenCL: A parallel programming standard for heterogeneous computing systems. John E. Stone, David Gohara, and Guochun Shi. Computing in Science and Engineering, 12:66-73, 2010.
- An Asymmetric Distributed Shared Memory Model for Heterogeneous Parallel Systems. Isaac Gelado, John E. Stone, Javier Cabezas, Sanjay Patel, Nacho Navarro, and Wen-mei W. Hwu. ASPLOS '10: Proceedings of the 15th International Conference on Architectural Support for Programming Languages and Operating Systems, pp. 347-358, 2010.
- GPU Clusters for High Performance Computing. Volodymyr Kindratenko, Jeremy Enos, Guochun Shi, Michael Showerman, Galen Arnold, John E. Stone, James Phillips, Wen-mei Hwu. Cluster Computing and Workshops, 2009. CLUSTER '09. IEEE International Conference on. pp. 1-8, Aug. 2009.
- Long time-scale simulations of in vivo diffusion using GPU hardware.
Elijah Roberts, John E. Stone, Leonardo Sepulveda, Wen-mei W. Hwu, and Zaida Luthey-Schulten.
In IPDPS '09: Proceedings of the 2009 IEEE International Symposium on Parallel & Distributed Processing, pp. 1-8, 2009
Publications Database - Probing biomolecular machines with graphics processors. James C. Phillips and John E. Stone. Communications of the ACM, 52:34-41, 2009.
- High performance computation and interactive display of molecular orbitals on GPUs and multi-core CPUs. John E. Stone, Jan Saam, David J. Hardy, Kirby L. Vandivort, Wen-mei W. Hwu, and Klaus Schulten. In Proceedings of the 2nd Workshop on General-Purpose Processing on Graphics Processing Units, ACM International Conference Proceeding Series, volume 383, pp. 9-18, New York, NY, USA, 2009. ACM.
- Multilevel summation of electrostatic potentials using graphics processing units. David J. Hardy, John E. Stone, and Klaus Schulten. Journal of Parallel Computing, 35:164-177, 2009.
- Adapting a message-driven parallel application to GPU-accelerated clusters. James C. Phillips, John E. Stone, and Klaus Schulten. In SC '08: Proceedings of the 2008 ACM/IEEE Conference on Supercomputing, Piscataway, NJ, USA, 2008. IEEE Press. (9 pages).
- GPU acceleration of cutoff pair potentials for molecular modeling applications. Christopher I. Rodrigues, David J. Hardy, John E. Stone, Klaus Schulten, and Wen-mei W. Hwu. In CF'08: Proceedings of the 2008 Conference on Computing Frontiers, pp. 273-282, New York, NY, USA, 2008. ACM.
- GPU computing. John D. Owens, Mike Houston, David Luebke, Simon Green, John E. Stone, and James C. Phillips. Proceedings of the IEEE, 96:879-899, 2008.
- Continuous fluorescence microphotolysis and correlation spectroscopy using 4Pi microscopy. Anton Arkhipov, Jana Hüve, Martin Kahms, Reiner Peters, and Klaus Schulten. Biophysical Journal, 93:4006-4017, 2007.
- Accelerating molecular modeling applications with graphics processors. John E. Stone, James C. Phillips, Peter L. Freddolino, David J. Hardy, Leonardo G. Trabuco, and Klaus Schulten. Journal of Computational Chemistry, 28:2618-2640, 2007.
Presentations
- Bringing State-of-the-Art GPU Accelerated Molecular Modeling Tools to the Research Community, GPU Technology Conference, San Jose, CA (03/20/2019)
- Interactive High Fidelity Biomolecular and Cellular Visualization with RTX Ray Tracing APIs, GPU Technology Conference, San Jose, CA (03/20/2019)
- Molecular Visualization and Simulation in VR, Indiana University, IN (3/6/2019)
- Adapting Scientific Software and Designing Algorithms for Next Generation GPU Computing Platforms, SIAM Conference on Computational Science and Engineering, Spokane, WA (02/27/2019)
- Challenges for Analysis and Visualization of Atomic-Detail Simulations of Minimal Cells, SIAM Conference on Computational Science and Engineering, Spokane, WA (02/26/2019)
- Optimizing NAMD and VMD for the IBM Power9 Platform, IBM Power User's Group, Supercomputing 2018, Dallas, TX (11/15/2018)
- NVIDIA HPC and University Computing Panel, Supercomputing 2018, Dallas, TX (11/14/2018)
- Using AWS EC2 GPU Instances for Computational Microscopy and Biomolecular Simulation, Amazon AWS Theater, Supercomputing 2018, Dallas, TX (11/14/2018)
- Interactivity in HPC BoF, Supercomputing 2018, Dallas, TX (11/13/2018)
- Putting High-Octane GPU-Accelerated Molecular Modeling Tools in the Hands of Scientists, NVIDIA Theater, Supercomputing 2018, Dallas, TX (11/13/2018)
- Blue Waters GPU Hackathon, National Center for Supercomputing Applications, University of Illinois, Urbana, IL (9/10/2018)
- GPU-Accelerated OptiX Ray Tracing for Scientific Visualization, NVIDIA Theater, Siggraph 2018, Vancouver BC, Canada (8/15/2018)
- VMD: Immersive Molecular Visualization with High-Fidelity Ray Tracing, Immersive Visualization for Science and Research, Siggraph 2018, Vancouver BC, Canada (8/13/2018)
- Argonne Training Program on Exascale Computing, Q Center, St. Charles, IL (8/9/2018)
- Preparing and Analyzing Large Molecular Simulations with VMD, Modeling Supra-molecular Structures with LAMMPS, Temple University, Philadelphia, PA (7/9 to 7/13, 2018)
- Interactive HPC Requirements, Challenges, and Solutions for Cutting Edge Molecular Simulation Science Campaigns, First Workshop on Interactive High-Performance Computing, ISC 2018, Frankfurt, Germany (6/28/2018)
- Visualizing the Atomic Detail Dynamics of Biomolecular Complexes in Our Compute-Rich but I/O-Constrained Future,
- Keynote: Visualization Challenges and Opportunities Posed by Petascale Molecular Dynamics Simulations, Workshop on Molecular Graphics and Visual Analysis of Molecular Data, EuroVis 2018, Brno, Czech Republic (6/4/2018)
- Advances in Biomolecular Simulation with NAMD and VMD, Pawsey Supercomputing Centre, Perth, Australia (4/19/2018)
- Pawsey GPU Hackathon, Pawsey Supercomputing Centre, Perth, Australia (4/16 to 4/20, 2018)
- VMD: Biomolecular Visualization from Atoms to Cells Using Ray Tracing, Rasterization, and VR, GPU Technology Conference, San Jose, CA (3/29/2018)
- Using Accelerator Directives to Adapt Science Applications for State-of-the-Art HPC Architectures, GPU Technology Conference, San Jose, CA (3/27/2018)
- Accelerating Molecular Modeling on Desktop and Pre-Exascale Supercomputers, GPU Technology Conference, San Jose, CA (3/26/2018)
- Optimizing HPC Simulation and Visualization Code Using NVIDIA Nsight Systems, GPU Technology Conference, San Jose, CA (3/29/2018)
- Interactive Supercomputing for State-of-the-Art Biomolecular Simulation, Interactivity in Supercomputing Birds of Feather, Supercomputing 2017, Denver, CO (11/14/2017)
- Using Accelerator Directives to Adapt Science Applications for State-of-the-Art HPC Architectures, Keynote Presentation, WAACPD'17: Fourth Workshop on Accelerator Programming Using Directives, Supercomputing 2017, Denver, CO (11/13/2017)
- VMD: GPU-Accelerated Analysis of Biomolecular and Cellular Simulations, Cray Analytics Symposium (remote webinar presentation), Radisson Blu MOA, Bloomington, MN (05/26/2017)
- GPUs Unleashed: Analysis of Petascale Molecular Dynamics Simulations with VMD, GPU Technology Conference, San Jose, CA (05/10/2017)
- Cutting Edge OptiX Ray Tracing Techniques for Visualization of Biomolecular and Cellular Simulations in VMD, GPU Technology Conference, San Jose, CA (05/09/2017)
- Turbocharging VMD Molecular Visualizations with State-of-the-Art Rendering and VR Technologies, GPU Technology Conference, San Jose, CA (05/09/2017)
- Visualizing Biomolecules in VMD, Hands-on Workshop on Computational Biophysics, NIH Center for Macromolecular Modeling and Bioinformatics, Beckman Institute, University of Illinois, Urbana, IL (4/18/2017).
- VMD: Preparation and Analysis of Molecular and Cellular Simulations, HPC for Life Sciences PRACE Spring School 2017, Stockholm Sweden (4/10/2017).
- Harnessing GPUs to Probe Biomolecular Machines at Atomic Detail, NVIDIA GPU Technology Theater, Supercomputing 2016, Supercomputing 2016, Salt Lake City, UT (11/16/2016).
- Visualization and Analysis of Biomolecular Complexes on Upcoming KNL-based HPC Systems, Intel HPC Developers Conference, Supercomputing 2016, Salt Lake City, UT (11/13/2016).
- Immersive Molecular Visualization with Omnidirectional Stereoscopic Ray Tracing and Remote Rendering, High Performance Data Analysis and Visualization Workshop, Chicago (5/23/2016).
- High Performance Molecular Visualization: In-Situ and Parallel Rendering with EGL, High Performance Data Analysis and Visualization Workshop, Chicago (5/23/2016)
- Evaluation of Emerging Energy-Efficient Heterogeneous Computing Platforms for Biomolecular and Cellular Simulation Workloads, 25th International Heterogeneity in Computing Workshop, Chicago (5/23/2016)
- VMD+OptiX: Streaming Interactive Ray Tracing from Remote GPU Clusters to Your VR Headset, GPU Technology Conference, San Jose, CA (4/06/2016)
- VMD: Interactive Molecular Ray Tracing with NVIDIA OptiX, GPU Technology Conference, San Jose, CA (4/06/2016)
- VMD: Petascale Molecular Visualization and Analysis with Remote Video Streaming, GPU Technology Conference, San Jose, CA (4/05/2016)
- VMD: enabling preparation, visualization, and analysis of petascale and pre-exascale molecular dynamics simulations, Armadillo B453 R1001, Lawrence Livermore National Laboratory, Livermore, CA (1/5/2016)
- VMD+OptiX: Bringing Interactive Molecular Ray Tracing from Remote GPU Clusters to your VR Headset, NVIDIA Theater, Supercomputing 2015, Austin, TX (11/18/2015)
- Chemical Visualization of Human Pathogens : The Retroviral Capsids, Visualization and Data Analytics Showcase, Supercomputing 2015, Austin, TX (11/18/2015)
- Frontiers of Molecular Visualization: Interactive Ray Tracing, Panoramic Displays, VR HMDs, and Remote Visualization, VisTech Workshop, Supercomputing 2015, Austin, TX (11/15/2015)
- VMD: Interactive Publication-Quality Molecular Ray Tracing with OptiX, NVIDIA Best of GTC Theater, Siggraph 2015, Los Angeles, CA (8/12/2015)
- VMD: Immersive Molecular Visualization and Interactive Ray Tracing for Domes, Panoramic Theaters, and Head Mounted Displays, BOF: Immersive Visualization for Science and Research, Siggraph 2015, Los Angeles, CA (8/10/2015)
- Workshop on Accelerated High-Performance Computing in Computational Sciences (SMR 2760), Abdus Salam International Centre for Theoretical Physics, Miramare - Trieste, Italy (05/27/2015)
- Panel: Data Relevancy, Locality, Diversity & More, NCSA Private Sector Program Annual Meeting, (05/06/2015)
- GPGPU 2015 Advanced Methods for Computing with CUDA, University of Cape Town, South Africa (4/20/2015)
- Proteins and Mesoscale Data: Visualization of Molecular Dynamics, Visualizing Biological Data (VIZBI) 2015 Conference, Broad Institute of MIT and Harvard, Cambridge, MA (03/26/2015)
- Publication-Quality Ray Tracing of Molecular Graphics with OptiX, GPU Technology Conference (03/19/2015)
- Attacking HIV with Petascale Molecular Dynamics Simulations on Titan and Blue Waters, GPU Technology Conference (3/18/2015)
- Guest Presentation: Integrating OptiX in VMD (in David McAllister's Innovations in OptiX presentation), GPU Technology Conference (03/18/2015)
- VMD: Visualization and Analysis of Biomolecular Complexes with GPU Computing, GPU Technology Conference (03/18/2015)
- Visualization of Energy Conversion Processes in a Light Harvesting Organelle at Atomic Detail, Visualization and Data Analytics Showcase, Supercomputing 2014, New Orleans, LA (11/19/2014)
- GPU-Accelerated Analysis of Large Biomolecular Complexes, NVIDIA Booth, Supercomputing 2014, New Orleans, LA (11/18/2014)
- GPU-accelerated Visualization and Analysis of Petascale Molecular Dynamics Simulations, Integrated Imaging Initiative Workshop: Tomography and Ptychography, Argonne National Laboratory (9/30/2014)
- GPU-accelerated Visualization and Analysis of Petascale Molecular Dynamics Simulations, Research and Technology Development Conference, Missouri University of Science and Technology (9/16/2014)
- Programming for Hybrid Architectures Today and in the Future, LSU Third Annual HPC User Symposium, Louisiana State University (6/5/2014)
- GPU-Accelerated Visualization and Analysis of Biomolecular Complexes, Oxford University, United Kingdom (5/12/2014)
- GPU-Accelerated Analysis and Visualization of Large Structures Solved by Molecular Dynamics Flexible Fitting, Faraday Discussion 169: Molecular Simulations and Visualization, University of Nottingham, United Kingdom (5/8/2014)
- Petascale molecular ray tracing: accelerating VMD/Tachyon with OptiX, GPU Technology Conference (03/27/2014)
- Fighting HIV with CUDA, CCoE Achievement Award, GPU Technology conference (03/25/2014)
- Visualization and analysis of petascale molecular simulations with VMD, GPU Technology Conference (03/25/2014)
- Using GPUs to Supercharge Visualization and Analysis of Molecular Dynamics Simulations with VMD, NVIDIA GTC Express (02/25/2014)
- Experiences Developing and Maintaining Scientific Applications on GPU-Accelerated Platforms, Indiana University, IUPUI Campus, Indianapolis, IN (01/24/2014)
- Fighting HIV with GPU-Accelerated Petascale Computing, Supercomputing 2013, Denver, CO (11/19/2013)
- GPU-Accelerated Molecular Visualization on Petascale Supercomputing Platforms, UltraVis'13: Eighth Ultrascale Visualization Workshop, Denver, CO (11/17/2013)
- Visualization and Analysis of Petascale Molecular Dynamics Simulations, San Diego Supercomputer Center, University of California San Diego (11/5/2013)
- Experiences Developing and Maintaining Scientific Applications on GPU-Accelerated Platforms, Indiana University, Bloomington, IN (10/2/2013)
- Interactive Molecular Visualization and Analysis with GPU Computing, Fall National Meeting of the American Chemical Society, Indianapolis, IN (9/11/2013)
- Early Experiences Scaling VMD Molecular Visualization and Analysis Jobs on Blue Waters, XSEDE Extreme Scaling Workshop, Boulder, CO (8/15/2013)
- GPU-Accelerated Molecular Visualization and Analysis with VMD, Midwest Theoretical Chemistry Conference, Urbana, IL, (5/31/2013)
- VMD: GPU-Accelerated Visualization and Analysis of Petascale Molecular Dynamics Simulations, GPU Technology Conference, San Jose, CA (3/20/2013)
- Petascale Molecular Dynamics Simulations on Titan and Blue Waters, GPU Technology Conference, San Jose, CA (3/19/2013)
- Keynote: Broadening the Use of Scalable Kernels in NAMD/VMD, VSCSE Many-core Processors, NCSA, University of Illinois (08/16/2012)
- GPU-Accelerated Analysis of Petascale Molecular Dynamics Simulations with VMD, Scalable Software for Scientific Computing Workshop, University of Notre Dame (6/11/2012)
- Visualization of petascale molecular dynamics simulations, Imaging at Illinois, The Next Generation: Computational Imaging and Visualization, Beckman Institute, University of Illinois (6/1/2012)
- Future Direction with NAMD, NAIS: State-of-the-Art Algorithms for Molecular Dynamics, Edinburgh, Scotland (5/24/2012)
- Demonstration: Using NAMD, NAIS: State-of-the-Art Algorithms for Molecular Dynamics, Edinburgh, Scotland (5/24/2012)
- Multilevel Summation Method in NAMD and VMD, NAIS: State-of-the-Art Algorithms for Molecular Dynamics, Edinburgh, Scotland (5/24/2012)
- Generalized Born Implicit Solvent Algorithm in NAMD, NAIS: State-of-the-Art Algorithms for Molecular Dynamics, Edinburgh, Scotland (5/24/2012)
- Short-Range (Non-Bonded) Interactions in NAMD, NAIS: State-of-the-Art Algorithms for Molecular Dynamics, Edinburgh, Scotland (5/24/2012)
- NAMD Algorithms and HPC Functionality, NAIS: State-of-the-Art Algorithms for Molecular Dynamics, Edinburgh, Scotland (5/24/2012)
- Analysis and Visualization Algorithms in VMD, NAIS: State-of-the-Art Algorithms for Molecular Dynamics, Edinburgh, Scotland (5/24/2012)
- In-Situ Visualization and Analysis of Petascale Molecular Dynamics Simulations with VMD, Accelerated HPC Symposium, San Jose, CA (5/17/20120
- High Performance Molecular Visualization and Analysis on GPUs, GPU Technology Conference, San Jose, CA (5/16/2012)
- Petascale Molecular Dynamics Simulations on GPU-Accelerated Supercomputers, GPU Technology Conference, San Jose, CA (5/16/2012)
- Scalable Molecular Dynamics with NAMD, Accelerating Computational Science Symposium 2012 (ACSS 2012), Oak Ridge National Laboratory (3/30/2012)
- Molecular Dynamics Simulations of Biomolecules on GPUs Using the Multilevel Summation Method, SIAM Conference on Computational Science and Engineering, Reno, NV (2/28/2011)
- Faster, Cheaper, Better: Biomolecular Simulation with NAMD, VMD, and CUDA, NVIDIA Booth, Supercomputing 2010, New Orleans, LA (11/16/2010)
- High Performance Computing with CUDA Case Study: Heterogeneous GPU Computing for Molecular Modeling, CUDA Tutorial, Supercomputing 2010, New Orleans, LA (11/14/2010)
- GPU and the Computational Microscope, GPU Technology Conference (09/22/2010)
- NAMD, CUDA, and Clusters: Taking GPU Molecular Dynamics Beyond the Desktop, GPU Technology Conference (09/23/2010)
- High Performance Molecular Simulation, Visualization, and Analysis on GPUs, GPU Technology Conference (09/22/2010)
- Simulating Biomolecules on GPUs with the Multilevel Summation Method, Oak Ridge National Laboratory (09/17/2010)
- High Performance Molecular Simulation, Visualization, and Analysis on GPUs, Oak Ridge National Laboratory (09/16/2010)
- Faster, Cheaper, and Better Science: Molecular Modeling on GPUs, Fall National Meeting of the American Chemical Society, Boston, MA (08/22/2010)
- OpenCL: Molecular Modeling on Heterogeneous Computing Systems, Fall National Meeting of the American Chemical Society, Boston, MA (08/22/2010)
- Quantifying the Impact of GPUs on Performance and Energy Efficiency in HPC Clusters, The Work in Progress in Green Computing(WIPGC), Chicago, IL (08/17/2010)
- Using GPUs to compute the multilevel summation of electrostatic forces, Multiscale Molecular Modeling Conference, Edinburgh, Scotland (07/02/2010)
- Molecular Visualization and Analysis on GPUs, Symposium on Application of GPUs in Chemistry and Materials Science, University of Pittsburgh (06/29/2010)
- Accelerating Biomolecular Modeling with CUDA and GPU Clusters, Accelerated Computing Conference, Tokyo, Japan (01/28/2010)
- An Introduction to OpenCL, GPUComputing.net Webinar (12/10/2009)
- Accelerating Molecular Modeling Applications with GPU Computing, Exhibition, Supercomputing 2009, Portland, OR (11/18/2009)
- OpenCL for Molecular Modeling Applications: Early Experiences, OpenCL BOF, Supercomputing 2009, Portland, OR (11/18/2009)
- An Introduction to OpenCL, IACAT/CCOE GPU Brown Bag Forum, University of Illinois (10/21/2009)
- High Performance Molecular Visualization and Analysis with GPU Computing, Beckman Institute Forum for Imaging and Visualization, University of Illinois (10/20/2009)
- Using GPU Computing to Accelerate Molecular Modeling Applications, CECAM Workshop, "Algorithmic Re-Engineering for Modern Non-Conventional Processing Units," CECAM-USI, Lugano, Switzerland (10/2/2009)
- GPU Accelerated Visualization and Analysis in VMD and Recent NAMD Developments, GPU Technology Conference, San Jose, CA (10/1/2009)
- Multilevel Summation of Electrostatic Potentials Using GPUs, Purdue University, (09/09/2009)
- Multidisciplinary Panel, VSCSE: Many-Core Processors for Science and Engineering Applications, NCSA (8/10/2008)
- GPU Accelerated Visualization and Analysis in VMD, Center for Molecular Modeling, University of Pennsylvania, (6/9/2009)
- Keynote: Accelerating Molecular Modeling Applications with GPU Computing, Second Sharcnet Symposium on GPU and Cell Computing, University of Waterloo (5/20/2009)
- Experiences with Multi-GPU Acceleration in VMD, Path to Petascale: Adapting GEO/CHEM/ASTRO Applications for Accelerators and Accelerator Clusters, NCSA (4/2/2009)
- Experience with NAMD on GPU-accelerated clusters, Path to Petascale: Adapting GEO/CHEM/ASTRO Applications for Accelerators and Accelerator Clusters, NCSA (4/2/2009)
- High Performance Computation and Interactive Display of Molecular Orbitals on GPUs and Multi-core CPUs, Second Workshop on General-Purpose Processing on Graphics Processing Units, Washington D.C. (3/8/2009)
- Adapting a Message-Driven Parallel Application to GPU-Accelerated Clusters, IACAT Accelerator Workshop, NCSA (1/23/2009)
- High Performance Computation and Interactive Display of Molecular Orbitals on GPUs and Multi-core CPUs, IACAT Accelerator Workshop, NCSA (1/23/2009)
- Adapting a Message-Driven Parallel Application to GPU-Accelerated Clusters, SC2008, Austin TX (11/18/2008)
- GPU Computing, Cape Linux Users Group, South Africa (10/28/2008)
- Accelerating Molecular Modeling Applications with Graphics Processors, Computer Science Department, University of Cape Town, South Africa (10/23/2008)
- Accelerating Computational Biology by 100x Using CUDA, NVISION 2008 (8/26/2008)
- GPU Acceleration of Cutoff Pair Potentials for Molecular Modeling Applications, ACM Computing Frontiers 2008 (5/7/2008)
- GPU Acceleration of Molecular Modeling Applications, Linux Clusters Institute Conference (5/1/2008)
- Accelerating Molecular Modeling Applications with Graphics Processors, SIAM PP08, Minisymposium 8: Revolutionary Technologies for Acceleration of Emerging Petascale Applications - GPUs (3/12/2008)
- Accelerating NAMD with Graphics Processors, SIAM PP08, Minisymposium 10: Current Developments in High-Performance Molecular Dynamics Simulations - Part II of II (3/12/2008)
- GPU Acceleration of Scientific Applications Using CUDA, AstroGPU 2007, Institute for Advanced Study, Princeton NJ (11/09/2007)
- Visualization of Nano-Scale Structures, University of Texas Health Science Center at Houston (4/20/2006)
- VMD: Algorithms and Methods for Large Scale Biomolecular Visualization, San Diego Supercomputer Center (9/12/2005)
Class lectures, workshop materials, and sample source code:
- GPGPU Computing with CUDA Workshop, University of Cape Town, South Africa (4/29/2013)
- High Performance Computing with CUDA Case Study: Heterogeneous GPU Computing for Molecular Modeling, CUDA Tutorial, Supercomputing 2010, New Orleans, LA (11/14/2010)
- Workshop on GPU Programming for Molecular Modeling, Beckman Institute, Urbana, IL (08/06/2010)
- The OpenCL Programming Model, Part 1 Illinois UPCRC Summer School (07/23/2010)
- The OpenCL Programming Model, Part 2 Illinois UPCRC Summer School (07/23/2010)
- Application Performance Case Studies: Molecular Visualization and Analysis (ECE 498 AL Guest Lecture) (4/8/2010)
- High Performance Computing with CUDA Case Study: Molecular Modeling Applications, CUDA Tutorial, Supercomputing 2009, Portland, OR (11/15/2009)
- Biomolecular Modeling Applications of GPUs and CPU-Accelerated Clusters, CUDA Tutorial, IEEE Cluster 2009 (9/4/2009)
- Case Study - Accelerating Molecular Dynamics Experimentation, VSCSE: Many-Core Processors for Science and Engineering Applications, NCSA (8/13/2008)
- Application Performance Case Studies: Molecular Visualization and Analysis (ECE 498 AL1 Guest Lecture) (4/7/2009, 4/9/2009)
- Intro: Using CUDA on Multiple GPUs Concurrently (IACAT Brown Bag Forum) (2/24/2009)
- GPU Computing Case Study: Molecular Modeling Applications (ECE 598 SP Guest Lecture) (11/11/2008)
- Case Study - Accelerating Molecular Dynamics Experimentation, Accelerators for Science and Engineering Applications: GPUs and Multicore, (8/21/2008)
- "Accelerating Scientific Applications with GPUs", Workshop on Programming Massively Parallel Processors (PMPP) (7/10/2008)
- Tutorial: High Performance Computing with CUDA (International Supercomputing Conference 2008) (6/16/2008)
- Tutorial: High Performance Computing on GPUs with CUDA (Supercomputing 2007)
- Performance Case Studies: Ion Placement Tool, VMD (ECE 498 AL1 Guest Lecture) (10/15/2007)
- Performance Case Studies: Ion Placement Tool, VMD (ECE 498 AL Guest Lecture) (3/14/2007)
- ECE 498 AL class home page
- Sponsored ECE 498 student projects
Investigators
|
Collaborators
|
Our Research in the News
|



{kind=link}